CREUTZFELDT JAKOB DISEASE TSE PRION DISEASE UPDATE USA DECEMBER 2023
Tables of Cases Examined
National Prion Disease Pathology Surveillance Center Cases Examined¹
Updated quarterly.
Last updated on: September 2nd, 2023
Year Total Neuropath Referrals² Prion Disease Sporadic Genetic Iatrogenic vCJD
1999 & earlier 383 232 202 27 3 0
2000 145 102 90 12 0 0
2001 209 118 110 8 0 0
2002 241 144 124 18 2 0
2003 259 160 137 21 2 0
2004 315 180 163 16 0 1³
2005 330 179 157 21 1 0
2006 365 179 159 17 1 2⁴
2007 374 210 191 19 0 0
2008 384 221 205 16 0 0
2009 397 231 210 20 1 0
2010 402 247 219 28 0 0
2011 392 238 214 24 0 0
2012 413 244 221 23 0 0
2013 416 258 223 34 1 0
2014 355 208 185 21 1 1⁵
2015 401 262 243 19 0 0
2016 395 277 248 29 0 0
2017 375 266 247 18 0 0
2018 308 221 202 18 1 0
2019 434 281 259 22 0 0
2020 367 253 228 24 1 0
2021 344 248 224 22 0 0
2022 337 228 206 21 0 0
2023 193 114 101 8 0 0
TOTAL 8534 5301 4720 507 14 4
=====
Year CSF Only and RT-QuIC Positive
2015 140
2016 183
2017 227
2018 266
2019 310
2020 309
2021 331
2022 347
2023 240
TOTAL 2357
Listed based on the year of death or, if not available, on the year of referral;
Cases with suspected prion disease for which brain tissue was submitted;
Disease acquired in the United Kingdom;
Disease acquired in the United Kingdom in one case and in Saudi Arabia in the other;
Disease possibly acquired in a Middle Eastern or Eastern European country;
Includes 28 cases in which the diagnosis is pending (2 from 2022 and 31 from 2023), and 20 inconclusive cases;
Includes 8 (2 from 2021, 1 from 2022, and 5 from 2023) cases with type determination pending in which the diagnosis of vCJD has been excluded.
The sporadic cases include 4641 cases of sporadic Creutzfeldt-Jakob disease (sCJD), 88 cases of Variably Protease-Sensitive Prionopathy (VPSPr), and 39 cases of sporadic Fatal Insomnia (sFI).
Total does not include 312 Familial cases diagnosed by blood test only.
Lists number of patients (deceased and alive) who have had a positive RT-QuIC and no neuropath examination.
For a downloadable PDF version of our quarterly table, please click the link below:
TEXAS CJD STATISTICS WOEFULLY OUTDATED, 2012 to 2021...terry
Creutzfeldt-Jakob Disease Case Counts and Percentages by Year and Age Category (2012-2021)
Year <55 Years of Age % <55 Years of Age ≥ 55 Years of Age % ≥ 55 Years of Age Total Cases
2012 6 27% 16 73% 22
2013 1 7% 13 93% 14
2014 5 19% 22 81% 27
2015 2 10% 18 90% 20
2016 6 18% 27 82% 33
2017 3 12% 22 88% 25
2018 4 11% 32 89% 36
2019 4 9% 42 91% 46
2020 9 22% 32 78% 41
2021 1 2% 44 98% 45
Total 41 13% 268 87% 309
TEXAS CJD MAP
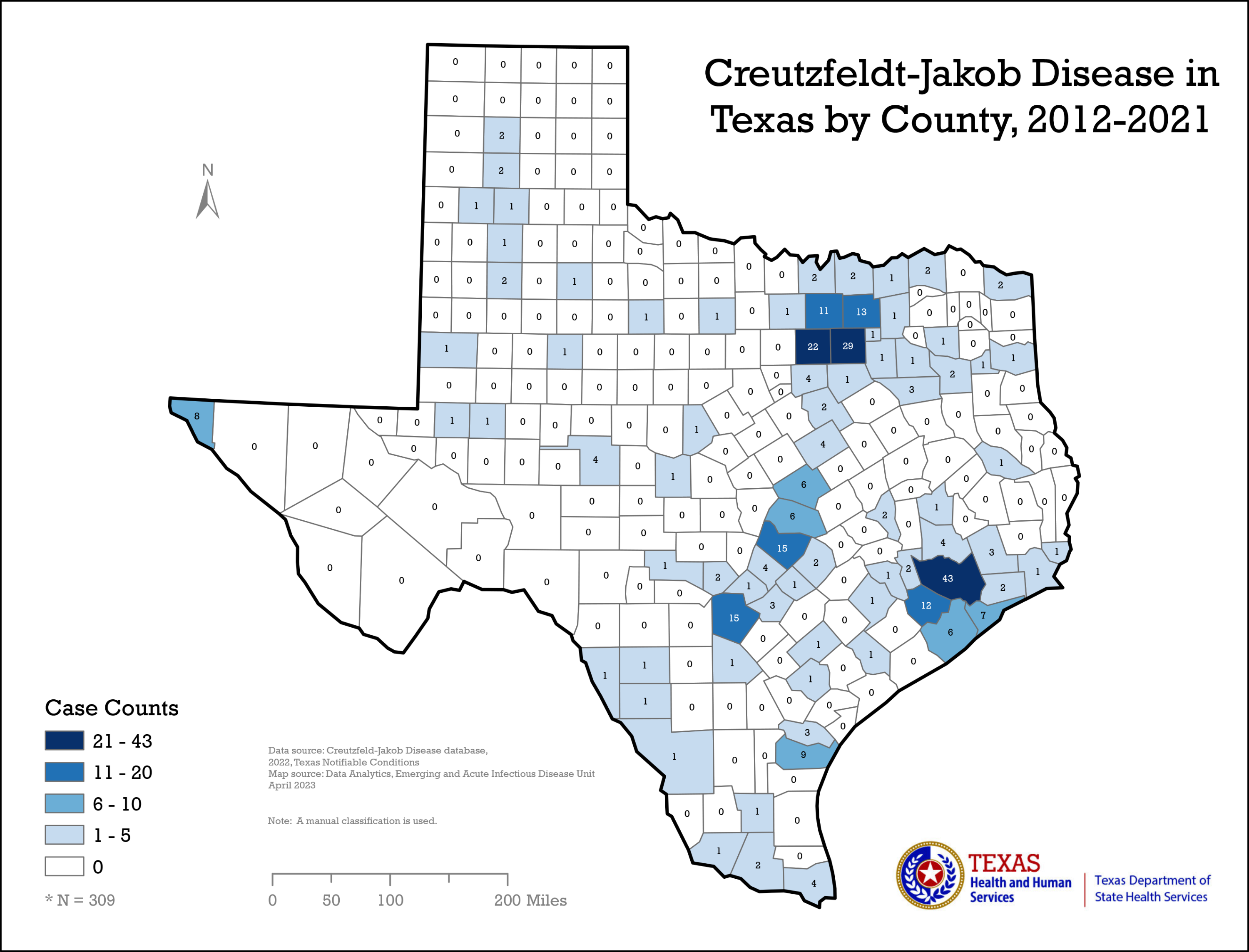{kind=link}
TEXAS CJD HISTORY
Research Letter
December 11, 2023
Change in Epidemiology of Creutzfeldt-Jakob Disease in the US, 2007-2020
Matthew A. Crane, BS1; Sameer Nair-Desai, BS2; Alison Gemmill, PhD3; et alJohn A. Romley, PhD4; John C. Probasco, MD5
Author Affiliations
1Johns Hopkins University School of Medicine, Baltimore, Maryland
2Department of Economics, Stanford University, Stanford, California
3Department of Population, Family and Reproductive Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland
4Leonard D. Schaeffer Center for Health Policy and Economics, University of Southern California, Los Angeles, California
5Department of Neurology, Johns Hopkins School of Medicine, Baltimore, Maryland
JAMA Neurol. Published online December 11, 2023. doi:10.1001/jamaneurol.2023.4678 Creutzfeldt-Jakob disease (CJD) is a rapidly progressive and universally fatal prion disease.1 Research on CJD in the US showed stable incidence from 1979 to 2006, though recent trends are not as well described.2 The incidence of sporadic CJD, the most common type, is higher among older patients.1,2 Due to aging populations worldwide, the epidemiology of CJD is evolving.3 We examined death certificate data from 2007 to 2020 to better understand recent US trends of CJD.
> Due to aging populations worldwide, the epidemiology of CJD is evolving.3
> We examined death certificate data from 2007 to 2020 to better understand recent US trends of CJD.
The role of environmental factors on sporadic Creutzfeldt-Jakob disease mortality: evidence from an age-period-cohort analysis
Eur J Epidemiol. 2023; 38(7): 757–764. Published online 2023 May 16. doi: 10.1007/s10654-023-01004-5 PMCID: PMC10276107PMID: 37191829
Received: 31 January 2023 / Accepted: 6 April 2023 / Published online: 16 May 2023 © The Author(s) 2023
The role of environmental factors on sporadic Creutzfeldt-Jakob disease mortality: evidence from an age-period-cohort analysis
Angéline Denouel1
· Jean-Philippe Brandel1,2
· Danielle Seilhean1 · Jean-Louis Laplanche3,4
· Alexis Elbaz5
· Stéphane Haik1,2
Abstract
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form of prion diseases. The causes of sCJD are still unknown and exogenous factors may play a role. Worldwide, the number of patients with sCJD has progressively increased over time. This increase can be partly explained by increasing life expectancy and better case ascertainment, but a true increase in the number of sCJD cases cannot be excluded. We estimated mortality rates from sCJD in France (1992–2016) and studied variation in mortality rates by age, period, and time.
We included all cases aged 45–89 years old who died with a probable/definite sCJD diagnosis based on the French national surveillance network. We used age-period-cohort (APC) Poisson regression models to study variation in mortality rates by sex, age, period, and time.
A total of 2475 sCJD cases aged 45–89 years were included. Mortality rates increased with age, reached a peak between 75 and 79 years, and decreased thereafter. Mortality rates were higher in women than men at younger ages and lower at older ages. The full APC model with a sex×age interaction provided the best fit to the data, thus in favour of sex, age, period, and cohort effects on mortality rates. In particular, mortality rates increased progressively with successive birth cohorts.
Based on 25 years of active surveillance in France, we show evidence for sex, age, period, and cohort effects on sCJD mortality. The identification of cohort effects suggests that environmental exposures may play a role in sCJD etiology.
Keywords Age-period-cohort model · Prion · Temporal trend · Sporadic Creutzfeldt-Jakob disease
snip...
Worldwide, the number of patients with sCJD appears to have progressively increased over time [13]. This increase can be partly explained by increasing life expectancy as well as by better case ascertainment due to improved diagnostic tests and awareness of the disease among clinicians. Indeed, a relationship between surveillance intensity and sCJD incidence has been shown [14]. It cannot be excluded, however, that an actual increase of sCJD cases has occurred, and this hypothesis can be examined using age-period-cohort (APC) models.
snip...
In this paper, we estimated mortality rates from sCJD in France over a 25-year period (1992–2016) based on data from the French national surveillance network.
snip...
The overall sCJD mortality rate was 4.58 per 1,000,000 person-years (95% CI=4.39–4.78) (Table S1).
snip...
Besides risk factors explored in case-control studies, the possibility of zoonotic risk factors remains a possibility that could account for an exogenous origin in some sCJD cases. Research on atypical forms of BSE (L-BSE, H-BSE) has revealed molecular similarities between the L-BSE strain and molecular subtypes of human sCJD, in particular the MV2 subtype [39]. Furthermore, L-BSE has been experimentally transmitted to non-human primates as efficiently as classical BSE responsible for vCJD in humans, and could be even more virulent [40–42]. The zoonotic risk associated with natural sheep scrapie has also been recently updated with the demonstration of an intracerebral transmission of scrapie to mice expressing the human prion protein during serial passages, as well as transmission of scrapie to primates. These observations highlight the possibility of a causal link between exposure to sheep scrapie and sCJD in some cases [43, 44]. A large increase in animal product consumption and the generalization of mechanically separated meat in developed countries over the last century may have contribute to increase the zoonotic prion pressure [45]. It would be of interest to observe the effect of safety measures implemented since the “mad cow crisis” to avoid population prion exposure on sCJD mortality in the next decades.
Prion 2023
Title: Diagnostic Journey of Patients with Creutzfeldt-Jakob Disease (CJD) in the United States: A RealWorld Evidence Study
Author list: Duncan Brown1 , Emily Kutrieb2 , Montserrat Vera Llonch1 , Rob Pulido1 , Anne Smith1 , Derek Weycker2 , Ellen Dukes2 , Brian S Appleby3-5
Affiliations: 1 Ionis Pharmaceuticals; 2Policy Analysis Inc. (PAI); 3National Prion Disease Pathology Surveillance Center; 4Case Western Reserve University; 5University Hospitals Cleveland Medical Center
Aims: Identification of clinical symptoms leading to a diagnosis of CJD from real-world evidence is limited. A new study using a United States (US) healthcare claims database was thus undertaken to address this evidence gap.
Materials and Methods: A retrospective cohort design and the Merative MarketScan Database (01/2012-12/2020) were employed. The study population comprised adults aged ≥18 years with ≥1 inpatient diagnosis or ≥2 outpatient diagnoses (≥3 days apart) of CJD, magnetic resonance imaging of the head or lumbar puncture, and no evidence of selected neurologic conditions after the last CJD diagnosis. Patients without healthcare coverage during the 12-month pre-diagnosis period were excluded; alternative pre-diagnosis periods (spanning 24 and 36 months, respectively) were also explored. Diagnostic journey was detailed based on diagnosis codes for selected symptoms and neurologic conditions during the pre-diagnosis period.
Results: Among the 61.8 million persons in the source population from 01/2013-12/2019, 215 CJD patients qualified for inclusion in the study population. CJD patients first presented with symptoms consistent with the diagnosis 5.0 (SD=4.0) months, on average, before the initial CJD diagnosis, and 80% had ≥3 symptoms, most commonly altered mental status (82%), gait/coordination disturbance (60%), and malaise/fatigue (44%). Most patients (63%) also had ≥1 differential (neurologic) diagnosis leading to the CJD diagnosis, most commonly cerebrovascular disease (49%), peripheral vertigo (11%), and Alzheimer’s disease (7%); mean duration from first differential diagnosis to initial CJD diagnosis was 2.4 (SD=3.1) months.
Conclusions: Study findings suggest that, in US clinical practice, CJD patients present with one or more clinical symptoms impacting motor, cognitive or other domains, and many are initially mis-diagnosed, prolonging the diagnostic journey. CJD should be considered in the differential diagnosis of those with rapidly progressing dementia or motor disturbance.
Funded by: Ionis Pharmaceuticals
Grant number: N/A
Acknowledgment: XXX
"Study findings suggest that, in US clinical practice, CJD patients present with one or more clinical symptoms impacting motor, cognitive or other domains, and many are initially mis-diagnosed, prolonging the diagnostic journey."
22 years ago;
2001 Singeltary on CJD
February 14, 2001
Diagnosis and Reporting of Creutzfeldt-Jakob Disease
Terry S. Singeltary, Sr
Author Affiliations
JAMA. 2001;285(6):733-734. doi:10-1001/pubs.JAMA-ISSN-0098-7484-285-6-jlt0214
To the Editor: In their Research Letter, Dr Gibbons and colleagues1 reported that the annual US death rate due to Creutzfeldt-Jakob disease (CJD) has been stable since 1985. These estimates, however, are based only on reported cases, and do not include misdiagnosed or preclinical cases. It seems to me that misdiagnosis alone would drastically change these figures. An unknown number of persons with a diagnosis of Alzheimer disease in fact may have CJD, although only a small number of these patients receive the postmortem examination necessary to make this diagnosis. Furthermore, only a few states have made CJD reportable. Human and animal transmissible spongiform encephalopathies should be reportable nationwide and internationally.
FRIDAY, JANUARY 15, 2021
CJD TSE Prion Questionnaire USA, UK, and the history there from, have you filled out this questionnaire?
if not, why not?
CJD TSE Prion Questionnaire USA, UK, Singeltary
CJD FOUNDATION Questionnaire
UK CJD Questionnaire
cjd questionnaire 1979
RE: re-Human Prion Diseases in the United States part 2 flounder replied to flounder on 02 Jan 2010 at 21:26 GMT I would kindly like to add to my initial concerns, something I brought up years ago, and I believe that still hold true today, more so even than when I first stated these concerns in 2003 ;
***> routine passive mortality CJD surveillance USA ?
***> THIS has been proven not to be very useful in the U.K.;
THE EPIDEMIOLOGY OF CJD RG WILL 1984 (182 PAGES)
snip...
One reason for this was the _inaccuracy_ in coding of cases correctly certified as CJD Coding is carried out by staff who are not medically qualified and it is not surprising that coding errors occur in the processing of large numbers of certificates. In 1982, 12,000 certificates per week were processed at the office of population censuses and surveys by 15 coders and 6 checkers (Alderson et al., 1983). The occurrence of both inter- and intra-observer coding errors has been described (Curb et al., 1983) and the _inaccuracies_ of BOTH certification and coding discovered in this study _support_ the introduction of a more accurate system of death certificates and a more detailed and specific coding system...
snip...
Draft Proposal For The Monitoring of Creutzfeldt-Kakob Disease 1989 Dr. R. Will
snip...
IDENTIFICATION OF CASES
Cases of CJD may be identified from death certificates, but this alone is unlikely to provide adequate monitoring. ERRORS are made in certification and diagnosis; in the Oxford study death certificates were obtained on a series of known confirmed cases and CJD was mentioned in only 66% of certificates. In another series of 175 certified cases, 42 patients were judged not to have suffered from CJD after examination of case notes (7)...
full text;
CJD Questionnaire
F. MEDICATIONS, has Subject taken any medications regularly, (if yes, record the date, name of the medication, the reason for taking it, and route of administration) prompt for prescription drugs, including insulin and type. Prompt for hormone therapy or nutritional supplements including oral contraceptives and hormone replacement therapy: Prompt for homeopathic/herbal therapy: Prompt for eye drops SUMMARY OF ABOVE RESPONSES; HAS THE SUBJECT BEEN EXPOSED TO ONE OF THE MEDICATIONS OF BOVINE OR OVINE ORIGIN, AND OR ANY DESICCATED ANIMAL ORIGIN? G. Has Subject ever been tested for allergy using needles? H. Has Subject ever received a treatment involving a course of injections? (If yes, record year, name of therapy, frequency, reason)
NOT to open up old wounds, but here is my 23 years of endeavors to get the USA to have a CJD Questionnaire for every family of a person whom died of cjd tse prion in the USA in every State, pertaining to real questions of all the potential routes of CJD in that questionnaire. seems i have failed terribly. there was great debate, much anguish, and i did take it personally, when others took credit for what i had been trying to get done. but this was long ago, and today the CJD Foundation seems to be working hard to change there old ways, and seem to be looking to find the routes of sporadic cjd as well. this is just that history, like it or not...kind regards, terry
THE MAKING OF THE USA CJD QUESTIONNAIRE
MONDAY, SEPTEMBER 11, 2023
Professor John Collinge on tackling prion diseases sCJD around 1 in 5000 deaths worldwide
“The best-known human prion disease is sporadic Creutzfeldt-Jakob disease (sCJD), a rapidly progressive dementia which accounts for around 1 in 5000 deaths worldwide.”
Singeltary sCJD
AS implied in the Inset 25 we must not _ASSUME_ that transmission of BSE to other species will invariably present pathology typical of a scrapie-like disease.
snip...
http://web.archive.org/web/20060307063542/http://www.bseinquiry.gov.uk/files/yb/1991/01/04004001.pdf
CANADA
Definite and probable CJD, 1998-2023
As of November 30, 2023
Year Sporadic Iatrogenic Genetic vCJD Total
1998 22 1 1 0 24
1999 27 2 3 0 32
2000 32 0 3 0 35
2001 27 0 3 0 30
2002 30 0 5 1 36
2003 27 1 1 0 29
2004 42 0 4 0 44
2005 42 0 2 0 44
2006 39 0 5 0 44
2007 35 0 4 0 39
2008 48 0 1 0 49
2009 48 0 5 0 53
2010 35 0 3 0 38
2011 46 0 4 1 51
2012 62 0 1 0 63
2013 50 0 1 0 51
2014 51 0 5 0 56
2015 44 0 8 0 52
2016 57 1 6 0 64
2017 82 0 5 0 87
2018 75 1 5 0 81
2019 76 0 3 0 79
2020 65 0 4 0 69
2021 60 0 3 0 63
2022 88 0 2 0 90
2023 52 0 1 0 53
Total 1263 6 86 2 1357
Cases with definite and probable diagnosis to date
SNIP...
2023, STILL NO MENTION OF VPSPr TSE Prion in Canada statistics...terry
PLEASE NOTE, Canada does not mention VPSPr Variably protease-sensitive prionopathy and you can read why here ;
WHY do some countries count vpspr as sporadic cjd tse prion, and some countries don't?
THIS problem must be addressed immediately imo.
WE have the USA classifying Variably protease-sensitive prionopathy (VPSPr) (formerly known as Protease Sensitive Prionopathy) as sporadic Creutzfeldt Jakob Disease sCJD, and we have Canada not even mentioning in on there statistics links, like vpspr does not even exist, so this is a problem for any valid surveillance imo. IN fact, personal communication from Canada Surveillance et al;
QUOTE;
''Well Terry, we have the data. We simply do not report it separately because we do not believe it has any specific epidemiologic significance, including zoonotic transmission (this opinion is shared unanimously by the international CJD surveillance community, and was established very quickly after the discovery of VPSPr). The key reason in my mind why the US system reports it – in a footnote to their sporadic CJD data – is that they discovered it, and want to follow up on it publicly to validate the reality of their finding scientifically (which is distinct from its significance).''
''The simple answer to your question is that we do not track VPSPr separately, as we view is as a form of sporadic CJD with an unusual phenotype but no specific epidemiological significance. Even the USA surveillance figures do not report it separately.''
end
Hell of a way for a surveillance system for any country to look for any suspect unusual zoonosis zoonotic disease from any mutated TSE Prion strain from any species. ...terry
WEDNESDAY, NOVEMBER 22, 2023
Creutzfeldt-Jakob Disease in Mexico (1990-2023)
CREUTZFELDT-JAKOB DISEASE IN THE UK (By Calendar Year)
REFERRALS OF SUSPECT CJD DEATHS OF DEFINITE AND PROBABLE CJD
Year Referrals Year Sporadic1
Iatrogenic Genetic2
vCJD Total Deaths
1990 [53]† 1990 28 5 0 - 33
1991 75 1991 31 1 4 - 36
1992 96 1992 45 2 6 - 53
1993 79 1993 36 4 7 - 47
1994 119 1994 53 1 9 - 63
1995 87 1995 35 4 5 3 47
1996 132 1996 40 4 6 10 60
1997 163 1997 59 6 7 10 82
1998 155 1998 64 3 5 18 90
1999 170 1999 62 6 2 15 85
2000 178 2000 48 1 3 28 80
2001 179 2001 58 4 6 20 88
2002 164 2002 73 0 5 17 95
2003 163 2003 79 5 7 18 109
2004 114 2004 50 2 6 9 67
2005 124 2005 67 4 13 5 89
2006 112 2006 68 1 9 5 83
2007 119 2007 63 2 11 5 81
2008 150 2008 84 5 6 2 97
2009 153 2009 78 2 8 3 91
2010 150 2010 85 3 6 3 97
2011 158 2011 91 4 14 5 114
2012 127 2012 92 5 12 0 109
2013 152 2013 108 2 10 1 121
2014 130 2014 100 3 13 0 116
2015 140 2015 105 0 4 0 109
2016 148 2016 118 1 6 1 126
2017 159 2017 122 0 12 0 134
2018 167 2018 137 2 12 0 151
2019 147 2019 128 1 7 0 136
2020 172 2020 135 1 8 0 144
2021 179 2021 133 0 6 0 139
2022 182 2022 150 0 8 0 158
2023 145 2023 113 1 3 0 117
Total
Referrals 4741 Total
Deaths
2738 85 246 178 3247
† Referral figure for 1990 is from 1 May onwards...*As at 4th December 2023
Summary of vCJD cases
Deaths from definite vCJD (confirmed): 123
Deaths from probable vCJD (without neuropathological confirmation): 55
Number of deaths from definite or probable vCJD (as above): 178
Number of definite/probable vCJD cases still alive: 0
Total number of definite or probable vCJD (dead and alive): 178
1 There are, in addition, a total of 20 cases of vPSPr (death in 1997(1 case), 2004(1), 2006(1), 2008(3), 2010(1), 2012(4), 2013(1), 2016(3), 2017(1), 2018(1), 2019(1), 2020(2)) not included in the above figures.
2 includes all genetic prion disease, including GSS
Case Report
An autopsy case of variably protease-sensitive prionopathy with Met/Met homogeneity at codon 129
Akiko Uchino, Yuko Saito, Saori Oonuma, Shigeo Murayama, Saburo Yagishita, Tetsuyuki Kitamoto, Kazuko Hasegawa
First published: 30 May 2023
Abstract
The typical clinical manifestations of sporadic Creutzfeldt-Jakob disease (sCJD) are rapid-progressive dementia and myoclonus. However, the diagnosis of atypical sCJD can be challenging due to its wide phenotypic variations. We report an autopsy case of variably protease-sensitive prionopathy (VPSPr) with Met/Met homogeneity at codon 129. An 81-year-old woman presented with memory loss without motor symptoms. Seventeen months after the onset, her spontaneous language production almost disappeared. Diffusion-weighted images (DWI) showed hyperintensity in the cerebral cortex while electroencephalogram (EEG) showed nonspecific change. 14-3-3 protein and real-time qualing-induced conversion (RT-QuIC) of cerebrospinal fluid were negative. She died at age 85, 3.5 years after the onset. Pathological investigation revealed spongiform change, severe neuronal loss, and gliosis in the cerebral cortex. Mild to moderate neuronal loss and gliosis were observed in the basal ganglia. PrP immunostaining revealed plaque-like, dotlike, and synaptic structures in the cerebral cortex and small plaque-like structures in the molecular layer of the cerebellum. Analysis of PRNP showed no pathogenic mutations, and Western blot examination revealed the lack of a diglycosylated band consistent with VPSPr. The present case, which is the first report on a VPSPr case in Japan, supports previously published evidence that VPSPr cases can present variable and nonspecific clinical presentations. Because a small number of VPSPr cases can show typical magnetic resonance imaging (MRI) change in sCJD. We should investigate the possibility of VPSPr in a differential diagnosis with atypical dementia that presented DWIs of high intensity in the cortex, even though 14-3-3 proteins and RT-QuIC are both negative. In addition, VPSPr cases can take a longer clinical course compared to that of sCJD, and long-term follow-up is important.
FRIDAY, OCTOBER 27, 2023
Prion disease features in Japan based on the national surveillance from 1999 to 2023
FRIDAY, OCTOBER 27, 2023
Geographic Diversity in the Incidence of Human Prion Diseases — China, 2006–2019
Unforeseen decrease of full-length prion protein in macaques exposed to prion contaminated blood products
Emmanuel COMOY, Nina JAFFRE, Jérôme DELMOTTE, Jacqueline MIKOL, and Jean Philippe DESLYS Commissariat à l’Energie Atomique, DRF/IBFJ/SEPIA, 18 Route du Panorama, 92265 Fontenay-aux-Roses, France.
Aims: The presence of prion infectivity in blood from patients affected by variant of Creutzfeldt-Jakob disease (v-CJD) questions the risk of its inter-human transmission through transfusion. We have previously described that several cynomolgus macaques experimentally exposed to prion-contaminated blood products developed c-BSE/v-CJD; however, after an exposure to low infectious doses, the vast majority of them developed an unexpected, fatal disease phenotype focused on spinal cord involvement which does not fulfill the classical diagnostic criteria of v-CJD, notably concerning the pathognomonic accumulation of abnormal prion protein. Here we aim to investigate the etiology and physiopathology of this original myelopathy.
Materials and Methods: CNS (brain and spinal cord) samples from myelopathic macaques were tested with different biochemical approaches in comparison to samples derived from either healthy animals or their counterparts exposed to different strains of prion diseases.
Results: Current conventional techniques failed to detect any accumulation of abnormal prion protein (PrPv-CJD) in the CNS of these myelopathic animals. Conversely, in their spinal cord we observed an alteration of their physiological cellular PrP pattern: PrP was not detectable under its full-length classical expression but mainly under its physiological terminal-truncated C1 fragment.
Conclusions: We here confirm the prion origin of this original syndrome, with a very specific biochemical signature linked to changes in PrP at the level of spinal cord lesions: contrary to what is classically described in prion diseases, host PrP is here altered in a form that is abnormally sensitive to degradation by cellular catabolism. This could provide the first experimental evidence of a link between loss of function of the cellular prion protein and the onset of disease. These observations open up new horizons in the field of prion diseases, which has hitherto been limited to pathologies associated with abnormal changes in cellular PrP towards highly structured conformations, with the possibility of unsuspected prion mechanisms/origins in certain neurodegenerative disorders.
Funded by: Financial support for the study was provided by the French National Research Agency (ANR).
Grant number: ANR-10-BLAN-133001 and BIOTECS2010-BloodSecur
Acknowledgement: We specially thank N. Lescoutra, A. Culeux, V. Durand, E. Correia, C. Durand and S. Jacquin for precious technical assistance
Saturday, February 2, 2019
CWD GSS TSE PRION SPINAL CORD, Confucius Ponders, What if?
REVIEW
***> In conclusion, sensory symptoms and loss of reflexes in Gerstmann-Sträussler-Scheinker syndrome can be explained by neuropathological changes in the spinal cord. We conclude that the sensory symptoms and loss of lower limb reflexes in Gerstmann-Sträussler-Scheinker syndrome is due to pathology in the caudal spinal cord. <***
***> The clinical and pathological presentation in macaques was mostly atypical, with a strong emphasis on spinal cord pathology.<***
***> The notion that CWD can be transmitted orally into both new-world and old-world non-human primates asks for a careful reevaluation of the zoonotic risk of CWD. <***
***> All animals have variable signs of prion neuropathology in spinal cords and brains and by supersensitive IHC, reaction was detected in spinal cord segments of all animals.<***
***> In particular the US data do not clearly exclude the possibility of human (sporadic or familial) TSE development due to consumption of venison. The Working Group thus recognizes a potential risk to consumers if a TSE would be present in European cervids.'' Scientific opinion on chronic wasting disease (II) <***
2023 TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY TSE PRION DISEASE IN THE USA ZOONOSIS
Detection of classical BSE prions in asymptomatic cows after inoculation with atypical/Nor98 scrapie
Marina Betancor, Belén Marín, Alicia Otero, Carlos Hedman, Antonio Romero, Tomás Barrio, Eloisa Sevilla, Jean-Yves Douet, Alvina Huor, Juan José Badiola, Olivier Andréoletti & Rosa Bolea * Veterinary Research volume 54, Article number: 89 (2023) Abstract
The emergence of bovine spongiform encephalopathy (BSE) prions from atypical scrapie has been recently observed upon experimental transmission to rodent and swine models. This study aimed to assess whether the inoculation of atypical scrapie could induce BSE-like disease in cattle. Four calves were intracerebrally challenged with atypical scrapie. Animals were euthanized without clinical signs of prion disease and tested negative for PrPSc accumulation by immunohistochemistry and western blotting. However, an emergence of BSE-like prion seeding activity was detected during in vitro propagation of brain samples from the inoculated animals. These findings suggest that atypical scrapie may represent a potential source of BSE infection in cattle.
Snip…
Further in vivo experiments challenging different mouse lines have been started in order to confirm the infectivity of the PMCA products obtained in this study. However, in conclusion, our findings show that the propagation of atypical scrapie in cattle leads to the emergence of BSE-like seeding activity.
This is a concerning issue with far-reaching implications for public health and food safety. The possibility of interspecies transmission of prion diseases and the emergence of new prion strains highlight the critical need for continued surveillance and monitoring of these diseases in both animal and human populations. Early detection of prion diseases is crucial, and highly sensitive detection techniques such as PMCA can play an important role in this regard.
Wednesday, May 24, 2023
WAHIS, WOAH, OIE, United States of America Bovine spongiform encephalopathy Immediate notification
FRIDAY, MAY 19, 2023
USDA Announces Atypical L-Type Bovine Spongiform Encephalopathy BSE Detection
SATURDAY, MAY 20, 2023
Tennessee State Veterinarian Alerts Cattle Owners to Disease Detection Mad Cow atypical L-Type BSE
ABOUT 2+ WEEKS BEFORE THE DETECTION OF BSE IN THE USA IN 2023, I WROTE THIS;
May 2, 2023, i submitted this to the USDA et al;
Docket No. APHIS–2023–0027 Notice of Request for Revision to and Extension of Approval of an Information Collection; National Veterinary Services Laboratories; Bovine Spongiform Encephalopathy Surveillance Program Singeltary Submission
ONLY by the Grace of God, have we not had a documented BSE outbreak, that and the fact the USDA et al are only testing 25K cattle for BSE, a number too low to find mad cow disease from some 28.9 million beef cows in the United States as of Jan. 1, 2023, down 4% from last year. The number of milk cows in the United States increased to 9.40 million. U.S. calf crop was estimated at 34.5 million head, down 2% from 2021. Jan 31, 2023.
ALL it would take is one BSE positive, yet alone a handful of BSE cases, this is why the Enhanced BSE was shut down, and the BSE testing shut down to 25k, and the BSE GBRs were replaced with BSE MRRs, after the 2003 Christmas Mad cow, the cow that stole Christmas, making it legal to trade BSE, imo.
Document APHIS-2023-0027-0001 BSE Singeltary Comment Submission
see full submission;
Price of TSE Prion Poker goes up substantially, all you cattle ranchers and such, better pay close attention here...terry
Transmission of the chronic wasting disease agent from elk to cattle after oronasal exposure
Justin Greenlee, Jifeng Bian, Zoe Lambert, Alexis Frese, and Eric Cassmann Virus and Prion Research Unit, National Animal Disease Center, USDA-ARS, Ames, IA, USA
Aims: The purpose of this study was to determine the susceptibility of cattle to chronic wasting disease agent from elk.
Conclusions: Cattle with the E211K polymorphism are susceptible to the CWD agent after oronasal exposure of 0.2 g of infectious material.
"Cattle with the E211K polymorphism are susceptible to the CWD agent after oronasal exposure of 0.2 g of infectious material."
=====end
Strain characterization of chronic wasting disease in bovine-PrP transgenic mice
Conclusions: Altogether, these results exhibit the diversity of CWD strains present in the panel of CWD isolates and the ability of at least some CWD isolates to infect bovine species. Cattle being one of the most important farming species, this ability represents a potential threat to both animal and human health, and consequently deserves further study.
"Altogether, these results exhibit the diversity of CWD strains present in the panel of CWD isolates and the ability of at least some CWD isolates to infect bovine species. Cattle being one of the most important farming species, this ability represents a potential threat to both animal and human health, and consequently deserves further study."
=====end
Experimental transmission of ovine atypical scrapie to cattle Experimental transmission of ovine atypical scrapie to cattle
Timm Konold, John Spiropoulos, Janet Hills, Hasina Abdul, Saira Cawthraw, Laura Phelan, Amy McKenna, Lauren Read, Sara Canoyra, Alba Marín-Moreno & Juan María Torres
Veterinary Research volume 54, Article number: 98 (2023)
Abstract
Classical bovine spongiform encephalopathy (BSE) in cattle was caused by the recycling and feeding of meat and bone meal contaminated with a transmissible spongiform encephalopathy (TSE) agent but its origin remains unknown. This study aimed to determine whether atypical scrapie could cause disease in cattle and to compare it with other known TSEs in cattle. Two groups of calves (five and two) were intracerebrally inoculated with atypical scrapie brain homogenate from two sheep with atypical scrapie. Controls were five calves intracerebrally inoculated with saline solution and one non-inoculated animal. Cattle were clinically monitored until clinical end-stage or at least 96 months post-inoculation (mpi). After euthanasia, tissues were collected for TSE diagnosis and potential transgenic mouse bioassay. One animal was culled with BSE-like clinical signs at 48 mpi. The other cattle either developed intercurrent diseases leading to cull or remained clinical unremarkable at study endpoint, including control cattle. None of the animals tested positive for TSEs by Western immunoblot and immunohistochemistry. Bioassay of brain samples from the clinical suspect in Ov-Tg338 and Bov-Tg110 mice was also negative. By contrast, protein misfolding cyclic amplification detected prions in the examined brains from atypical scrapie-challenged cattle, which had a classical BSE-like phenotype. This study demonstrates for the first time that a TSE agent with BSE-like properties can be amplified in cattle inoculated with atypical scrapie brain homogenate.
snip...
This is the first study in cattle inoculated with naturally occurring scrapie isolates that found the presence of prions resembling classical BSE in bovine brain although this was limited to detection by the ultrasensitive PMCA. The results from thermostability assay confirmed that the isolates were as thermoresistant as the BSE agent as proven in other studies [36, 48]. Previous PMCA studies with various British atypical scrapie isolates did not find any evidence of amplification [49, 50]. This may be explained by the use of ovine brain as substrate rather than brain from Bov-Tg110 mice, which may facilitate conversion to classical BSE prions.
Two hypotheses for prion strain propagation in cross-species transmission experiments have been proposed: conformational selection favours a particular strain conformation out of a mixture of conformations in a scrapie isolate whilst mutation results in the conformational shift of one conformation into another [51]. Following on from the study in mice [17], it has been subsequently suggested that classical BSE properties that arise in atypical scrapie isolates transmitted to cattle may be due to conformational mutation in a new host [52]. It does not confirm that the atypical scrapie agent is the origin of the classical BSE epidemic and further transmission studies would be required to see whether classical BSE can be generated.
Would PMCA applied to brains from cattle exposed to TSE agents other than classical BSE and atypical scrapie also produce a classical BSE-like molecular phenotype? The PMCA product obtained in the thermostability test using a thermosensitive classical scrapie control showed a profile unlike classical BSE. Atypical BSE has been linked to the origin of classical BSE because of its conversion into classical BSE following serial passages in wild-type mice (L-type BSE [11]) and bovine transgenic mice (H-type BSE [53]). Although we have not tested PMCA products of atypical BSE isolates as part of this study, there is no evidence that PMCA products from atypical BSE convert into classical BSE, at least for H-type BSE using bovine brain as substrate [54]. In fact, we were unable to propagate H-type BSE using the same methodology (S Canoyra, A Marín-Moreno, JM Torres, unpublished observation).
The study results support the decision to maintain the current ban on animal meal in feedstuffs for ruminants, particularly as atypical scrapie occurs world-wide, and eradication is unlikely for a sporadic disease.
In summary, experimental inoculation of cattle with the atypical scrapie agent may produce clinical disease indistinguishable from classical BSE, which cannot be diagnosed by conventional diagnostic tests, but prions can be amplified by ultrasensitive tests in both clinically affected and clinically unremarkable cattle, which reveal classical BSE-like characteristics. Further studies are required to assess whether a BSE-like disease can be confirmed by conventional tests, which may initially include a second passage in cattle.
Title: Transmission of atypical BSE: a possible origin of Classical BSE in cattle
Authors: Sandor Dudas1, Samuel James Sharpe1, Kristina Santiago-Mateo1, Stefanie Czub1, Waqas Tahir1,2, *
Affiliation: 1National and WOAH reference Laboratory for Bovine Spongiform Encephalopathy, Canadian Food inspection Agency, Lethbridge Laboratory, Lethbridge, Canada. 2Department of Biological Sciences, University of Lethbridge, Lethbridge, Alberta, Canada.
*Corresponding and Presenting Author: waqas.tahir@inspection.gc.ca
Background: Bovine spongiform encephalopathy (BSE) is a fatal neurodegenerative disease of cattle and is categorized into classical and atypical forms. Classical BSE (CBSE) is linked to the consumption of BSE contaminated feed whereas atypical BSE is considered to be spontaneous in origin. The potential for oral transmission of atypical BSE is yet to be clearly defined.
Aims: To assess the oral transmissibility of atypical BSE (H and L type) in cattle. Should transmission be successful, determine the biochemical characteristics and distribution of PrPSc in the challenge cattle.
Material and Methods: For oral transmission, calves were fed with 100 g of either H (n=3) or L BSE (n=3) positive brain material. Two years post challenge, 1 calf from each of the H and L BSE challenge groups exhibited behavioural signs and were euthanized. Various brain regions of both animals were tested by traditional and novel prion detection methods with inconclusive results. To detect infectivity, brain homogenates from these oral challenge animals (P1) were injected intra-cranially (IC) into steer calves. Upon clinical signs of BSE, 3/4 of IC challenged steer calves were euthanized and tested for PrPSc with ELISA, immunohistochemistry and immunoblot.
Results: After 6 years of incubation, 3/4 animals (2/2 steers IC challenged with brain from P1 L-BSE oral challenge and 1/2 steer IC challenged with brain from P1 H-BSE oral challenge) developed clinical disease. Analysis of these animals revealed high levels of PrPSc in their brains, having biochemical properties similar to that of PrPSc in C-BSE.
Conclusion: These results demonstrate the oral transmission potential of atypical BSE in cattle. Surprisingly, regardless of which atypical type of BSE was used for P1 oral challenge, PrPSc in the P2 animals acquired biochemical characteristics similar to that of PrPSc in C-BSE, suggesting atypical BSE as a possible origin of C-BSE in UK.
Presentation Type: Oral Presentation
Funded by: CFIA, Health Canada, Alberta Livestock and Meat Agency, Alberta Prion Research Institute
Grant Number: ALMA/APRI: 201400006, HC 414250
spontaneous/sporadic CJD in 85%+ of all human TSE, or spontaneous BSE in cattle, is a pipe dream, dreamed up by USDA/OIE et al, that has never been proven. let me repeat, NEVER BEEN PROVEN FOR ALL HUMAN OR ANIMAL TSE I.E. ATYPICAL BSE OR SPORADIC CJD! please see;
***Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility.***
Even if the prevailing view is that sporadic CJD is due to the spontaneous formation of CJD prions, it remains possible that its apparent sporadic nature may, at least in part, result from our limited capacity to identify an environmental origin.
OIE Conclusions on transmissibility of atypical BSE among cattle
Given that cattle have been successfully infected by the oral route, at least for L-BSE, it is reasonable to conclude that atypical BSE is potentially capable of being recycled in a cattle population if cattle are exposed to contaminated feed. In addition, based on reports of atypical BSE from several countries that have not had C-BSE, it appears likely that atypical BSE would arise as a spontaneous disease in any country, albeit at a very low incidence in old cattle. In the presence of livestock industry practices that would allow it to be recycled in the cattle feed chain, it is likely that some level of exposure and transmission may occur. As a result, since atypical BSE can be reasonably considered to pose a potential background level of risk for any country with cattle, the recycling of both classical and atypical strains in the cattle and broader ruminant populations should be avoided.
Annex 7 (contd) AHG on BSE risk assessment and surveillance/March 2019
34 Scientific Commission/September 2019
3. Atypical BSE
The Group discussed and endorsed with minor revisions an overview of relevant literature on the risk of atypical BSE being recycled in a cattle population and its zoonotic potential that had been prepared ahead of the meeting by one expert from the Group. This overview is provided as Appendix IV and its main conclusions are outlined below. With regard to the risk of recycling of atypical BSE, recently published research confirmed that the L-type BSE prion (a type of atypical BSE prion) may be orally transmitted to calves1 . In light of this evidence, and the likelihood that atypical BSE could arise as a spontaneous disease in any country, albeit at a very low incidence, the Group was of the opinion that it would be reasonable to conclude that atypical BSE is potentially capable of being recycled in a cattle population if cattle were to be exposed to contaminated feed. Therefore, the recycling of atypical strains in cattle and broader ruminant populations should be avoided.
4. Definitions of meat-and-bone meal (MBM) and greaves
The L-type BSE prion is much more virulent in primates and in humanized mice than is the classical BSE prion, which suggests the possibility of zoonotic risk associated with the L-type BSE prion
Consumption of L-BSE–contaminated feed may pose a risk for oral transmission of the disease agent to cattle.
Thus, it is imperative to maintain measures that prevent the entry of tissues from cattle possibly infected with the agent of L-BSE into the food chain.
Atypical L-type bovine spongiform encephalopathy (L-BSE) transmission to cynomolgus macaques, a non-human primate
Fumiko Ono 1, Naomi Tase, Asuka Kurosawa, Akio Hiyaoka, Atsushi Ohyama, Yukio Tezuka, Naomi Wada, Yuko Sato, Minoru Tobiume, Ken'ichi Hagiwara, Yoshio Yamakawa, Keiji Terao, Tetsutaro Sata
Affiliations expand
PMID: 21266763
Abstract
A low molecular weight type of atypical bovine spongiform encephalopathy (L-BSE) was transmitted to two cynomolgus macaques by intracerebral inoculation of a brain homogenate of cattle with atypical BSE detected in Japan. They developed neurological signs and symptoms at 19 or 20 months post-inoculation and were euthanized 6 months after the onset of total paralysis. Both the incubation period and duration of the disease were shorter than those for experimental transmission of classical BSE (C-BSE) into macaques. Although the clinical manifestations, such as tremor, myoclonic jerking, and paralysis, were similar to those induced upon C-BSE transmission, no premonitory symptoms, such as hyperekplexia and depression, were evident. Most of the abnormal prion protein (PrP(Sc)) was confined to the tissues of the central nervous system, as determined by immunohistochemistry and Western blotting. The PrP(Sc) glycoform that accumulated in the monkey brain showed a similar profile to that of L-BSE and consistent with that in the cattle brain used as the inoculant. PrP(Sc) staining in the cerebral cortex showed a diffuse synaptic pattern by immunohistochemistry, whereas it accumulated as fine and coarse granules and/or small plaques in the cerebellar cortex and brain stem. Severe spongiosis spread widely in the cerebral cortex, whereas florid plaques, a hallmark of variant Creutzfeldt-Jakob disease in humans, were observed in macaques inoculated with C-BSE but not in those inoculated with L-BSE.
see full text;
''H-TYPE BSE AGENT IS TRANSMISSIBLE BY THE ORONASAL ROUTE''
This study demonstrates that the H-type BSE agent is transmissible by the oronasal route. These results reinforce the need for ongoing surveillance for classical and atypical BSE to minimize the risk of potentially infectious tissues entering the animal or human food chains.
Comparing the Distribution of Ovine Classical Scrapie and Sporadic Creutzfeldt-Jakob Disease in Italy: Spatial and Temporal Associations (2002-2014)
Aim: This study aims to investigate potential spatial and temporal associations between Creutzfeldt-Jakob disease (CJD) in humans (2010-2014) and ovine classical scrapie (CS) (2002- 2006) in Italy, serving as a proxy for exposure.
Results: The analysis of data at the district level revealed no significant association. However, when considering aggregated regional data, all four models consistently indicated a statistically significant positive association, suggesting a higher incidence of the disease in humans as the regional incidence of sheep scrapie increased.
Conclusions: While the results are intriguing, it is important to acknowledge the inherent limitations of ecological studies. Nevertheless, these findings provide valuable evidence to formulate a hypothesis regarding the zoonotic potential of classical scrapie. Further investigations are necessary, employing specific designs such as analytical epidemiology studies, to test this hypothesis effectively.
=====
Transmission of Idiopathic human prion disease CJD MM1 to small ruminant mouse models (Tg338 and Tg501).
Results: No evidence of transmission was found on a first passage in Tg338 nor Tg501ovinized mice, but on second passage, 4/10 Tg338 mice succumbed to CJDMM1 (40% attack rate after 645 dpi) and 1/12 Tg501 mice (519dpi, 10 still alive). The remaining 2nd passages are still ongoing. Conclusions: In this poster, the neuropathological features of the resulting strain are discussed.
Transmission of scrapie prions to primate after an extended silent incubation period
*** In complement to the recent demonstration that humanized mice are susceptible to scrapie, we report here the first observation of direct transmission of a natural classical scrapie isolate to a macaque after a 10-year incubation period. Neuropathologic examination revealed all of the features of a prion disease: spongiform change, neuronal loss, and accumulation of PrPres throughout the CNS.
*** This observation strengthens the questioning of the harmlessness of scrapie to humans, at a time when protective measures for human and animal health are being dismantled and reduced as c-BSE is considered controlled and being eradicated.
*** Our results underscore the importance of precautionary and protective measures and the necessity for long-term experimental transmission studies to assess the zoonotic potential of other animal prion strains.
***Transmission data also revealed that several scrapie prions propagate in HuPrP-Tg mice with efficiency comparable to that of cattle BSE. While the efficiency of transmission at primary passage was low, subsequent passages resulted in a highly virulent prion disease in both Met129 and Val129 mice.
***Transmission of the different scrapie isolates in these mice leads to the emergence of prion strain phenotypes that showed similar characteristics to those displayed by MM1 or VV2 sCJD prion.
***These results demonstrate that scrapie prions have a zoonotic potential and raise new questions about the possible link between animal and human prions.
***Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility.***
Even if the prevailing view is that sporadic CJD is due to the spontaneous formation of CJD prions, it remains possible that its apparent sporadic nature may, at least in part, result from our limited capacity to identify an environmental origin.
O.05: Transmission of prions to primates after extended silent incubation periods: Implications for BSE and scrapie risk assessment in human populations
*** We recently observed the direct transmission of a natural classical scrapie isolate to macaque after a 10-year silent incubation period,
***with features similar to some reported for human cases of sporadic CJD, albeit requiring fourfold long incubation than BSE. Scrapie, as recently evoked in humanized mice (Cassard, 2014),
***is the third potentially zoonotic PD (with BSE and L-type BSE),
***thus questioning the origin of human sporadic cases.
==============
PRION 2015 CONFERENCE
PRION 2016 TOKYO
Saturday, April 23, 2016
SCRAPIE WS-01: Prion diseases in animals and zoonotic potential 2016
Prion. 10:S15-S21. 2016 ISSN: 1933-6896 1933-690X
WS-01: Prion diseases in animals and zoonotic potential
Transmission of the different scrapie isolates in these mice leads to the emergence of prion strain phenotypes that showed similar characteristics to those displayed by MM1 or VV2 sCJD prion.
These results demonstrate that scrapie prions have a zoonotic potential and raise new questions about the possible link between animal and human prions.
Title: Transmission of scrapie prions to primate after an extended silent incubation period)
*** In complement to the recent demonstration that humanized mice are susceptible to scrapie, we report here the first observation of direct transmission of a natural classical scrapie isolate to a macaque after a 10-year incubation period. Neuropathologic examination revealed all of the features of a prion disease: spongiform change, neuronal loss, and accumulation of PrPres throughout the CNS.
*** This observation strengthens the questioning of the harmlessness of scrapie to humans, at a time when protective measures for human and animal health are being dismantled and reduced as c-BSE is considered controlled and being eradicated.
*** Our results underscore the importance of precautionary and protective measures and the necessity for long-term experimental transmission studies to assess the zoonotic potential of other animal prion strains.
Tuesday, December 16, 2014
Evidence for zoonotic potential of ovine scrapie prions
Hervé Cassard,1, n1 Juan-Maria Torres,2, n1 Caroline Lacroux,1, Jean-Yves Douet,1, Sylvie L. Benestad,3, Frédéric Lantier,4, Séverine Lugan,1, Isabelle Lantier,4, Pierrette Costes,1, Naima Aron,1, Fabienne Reine,5, Laetitia Herzog,5, Juan-Carlos Espinosa,2, Vincent Beringue5, & Olivier Andréoletti1, Affiliations Contributions Corresponding author Journal name: Nature Communications Volume: 5, Article number: 5821 DOI: doi:10.1038/ncomms6821 Received 07 August 2014 Accepted 10 November 2014 Published 16 December 2014
Abstract
Although Bovine Spongiform Encephalopathy (BSE) is the cause of variant Creutzfeldt Jakob disease (vCJD) in humans, the zoonotic potential of scrapie prions remains unknown. Mice genetically engineered to overexpress the humanprion protein (tgHu) have emerged as highly relevant models for gauging the capacity of prions to transmit to humans. These models can propagate human prions without any apparent transmission barrier and have been used used to confirm the zoonotic ability of BSE. Here we show that a panel of sheep scrapie prions transmit to several tgHu mice models with an efficiency comparable to that of cattle BSE. ***The serial transmission of different scrapie isolates in these mice led to the propagation of prions that are phenotypically identical to those causing sporadic CJD (sCJD) in humans. ***These results demonstrate that scrapie prions have a zoonotic potential and raise new questions about the possible link between animal and human prions.
Subject terms: Biological sciences• Medical research At a glance
why do we not want to do TSE transmission studies on chimpanzees $
5. A positive result from a chimpanzee challenged severly would likely create alarm in some circles even if the result could not be interpreted for man. I have a view that all these agents could be transmitted provided a large enough dose by appropriate routes was given and the animals kept long enough. Until the mechanisms of the species barrier are more clearly understood it might be best to retain that hypothesis.
snip...
R. BRADLEY
1: J Infect Dis 1980 Aug;142(2):205-8
Oral transmission of kuru, Creutzfeldt-Jakob disease, and scrapie to nonhuman primates.
Gibbs CJ Jr, Amyx HL, Bacote A, Masters CL, Gajdusek DC.
Kuru and Creutzfeldt-Jakob disease of humans and scrapie disease of sheep and goats were transmitted to squirrel monkeys (Saimiri sciureus) that were exposed to the infectious agents only by their nonforced consumption of known infectious tissues. The asymptomatic incubation period in the one monkey exposed to the virus of kuru was 36 months; that in the two monkeys exposed to the virus of Creutzfeldt-Jakob disease was 23 and 27 months, respectively; and that in the two monkeys exposed to the virus of scrapie was 25 and 32 months, respectively. Careful physical examination of the buccal cavities of all of the monkeys failed to reveal signs or oral lesions. One additional monkey similarly exposed to kuru has remained asymptomatic during the 39 months that it has been under observation.
snip...
The successful transmission of kuru, Creutzfeldt-Jakob disease, and scrapie by natural feeding to squirrel monkeys that we have reported provides further grounds for concern that scrapie-infected meat may occasionally give rise in humans to Creutzfeldt-Jakob disease.
PMID: 6997404
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=6997404&dopt=Abstract
Recently the question has again been brought up as to whether scrapie is transmissible to man. This has followed reports that the disease has been transmitted to primates. One particularly lurid speculation (Gajdusek 1977) conjectures that the agents of scrapie, kuru, Creutzfeldt-Jakob disease and transmissible encephalopathy of mink are varieties of a single "virus". The U.S. Department of Agriculture concluded that it could "no longer justify or permit scrapie-blood line and scrapie-exposed sheep and goats to be processed for human or animal food at slaughter or rendering plants" (ARC 84/77)" The problem is emphasised by the finding that some strains of scrapie produce lesions identical to the once which characterise the human dementias"
Whether true or not. the hypothesis that these agents might be transmissible to man raises two considerations. First, the safety of laboratory personnel requires prompt attention. Second, action such as the "scorched meat" policy of USDA makes the solution of the acrapie problem urgent if the sheep industry is not to suffer grievously.
snip...
76/10.12/4.6
Nature. 1972 Mar 10;236(5341):73-4.
Transmission of scrapie to the cynomolgus monkey (Macaca fascicularis).
Gibbs CJ Jr, Gajdusek DC.
Nature 236, 73 - 74 (10 March 1972); doi:10.1038/236073a0
Transmission of Scrapie to the Cynomolgus Monkey (Macaca fascicularis)
C. J. GIBBS jun. & D. C. GAJDUSEK
National Institute of Neurological Diseases and Stroke, National Institutes of Health, Bethesda, Maryland
SCRAPIE has been transmitted to the cynomolgus, or crab-eating, monkey (Macaca fascicularis) with an incubation period of more than 5 yr from the time of intracerebral inoculation of scrapie-infected mouse brain. The animal developed a chronic central nervous system degeneration, with ataxia, tremor and myoclonus with associated severe scrapie-like pathology of intensive astroglial hypertrophy and proliferation, neuronal vacuolation and status spongiosus of grey matter. The strain of scrapie virus used was the eighth passage in Swiss mice (NIH) of a Compton strain of scrapie obtained as ninth intracerebral passage of the agent in goat brain, from Dr R. L. Chandler (ARC, Compton, Berkshire).
2001
Suspect symptoms
What if you can catch old-fashioned CJD by eating meat from a sheep infected with scrapie?
28 Mar 01
Like lambs to the slaughter
31 March 2001
by Debora MacKenzie Magazine issue 2284.
FOUR years ago, Terry Singeltary watched his mother die horribly from a degenerative brain disease. Doctors told him it was Alzheimer's, but Singeltary was suspicious. The diagnosis didn't fit her violent symptoms, and he demanded an autopsy. It showed she had died of sporadic Creutzfeldt-Jakob disease.
Most doctors believe that sCJD is caused by a prion protein deforming by chance into a killer. But Singeltary thinks otherwise. He is one of a number of campaigners who say that some sCJD, like the variant CJD related to BSE, is caused by eating meat from infected animals. Their suspicions have focused on sheep carrying scrapie, a BSE-like disease that is widespread in flocks across Europe and North America.
Now scientists in France have stumbled across new evidence that adds weight to the campaigners' fears. To their complete surprise, the researchers found that one strain of scrapie causes the same brain damage in mice as sCJD.
"This means we cannot rule out that at least some sCJD may be caused by some strains of scrapie," says team member Jean-Philippe Deslys of the French Atomic Energy Commission's medical research laboratory in Fontenay-aux-Roses, south-west of Paris. Hans Kretschmar of the University of Göttingen, who coordinates CJD surveillance in Germany, is so concerned by the findings that he now wants to trawl back through past sCJD cases to see if any might have been caused by eating infected mutton or lamb.
Scrapie has been around for centuries and until now there has been no evidence that it poses a risk to human health. But if the French finding means that scrapie can cause sCJD in people, countries around the world may have overlooked a CJD crisis to rival that caused by BSE.
Deslys and colleagues were originally studying vCJD, not sCJD. They injected the brains of macaque monkeys with brain from BSE cattle, and from French and British vCJD patients. The brain damage and clinical symptoms in the monkeys were the same for all three. Mice injected with the original sets of brain tissue or with infected monkey brain also developed the same symptoms.
As a control experiment, the team also injected mice with brain tissue from people and animals with other prion diseases: a French case of sCJD; a French patient who caught sCJD from human-derived growth hormone; sheep with a French strain of scrapie; and mice carrying a prion derived from an American scrapie strain. As expected, they all affected the brain in a different way from BSE and vCJD. But while the American strain of scrapie caused different damage from sCJD, the French strain produced exactly the same pathology.
"The main evidence that scrapie does not affect humans has been epidemiology," says Moira Bruce of the neuropathogenesis unit of the Institute for Animal Health in Edinburgh, who was a member of the same team as Deslys. "You see about the same incidence of the disease everywhere, whether or not there are many sheep, and in countries such as New Zealand with no scrapie." In the only previous comparisons of sCJD and scrapie in mice, Bruce found they were dissimilar.
But there are more than 20 strains of scrapie, and six of sCJD. "You would not necessarily see a relationship between the two with epidemiology if only some strains affect only some people," says Deslys. Bruce is cautious about the mouse results, but agrees they require further investigation. Other trials of scrapie and sCJD in mice, she says, are in progress.
People can have three different genetic variations of the human prion protein, and each type of protein can fold up two different ways. Kretschmar has found that these six combinations correspond to six clinical types of sCJD: each type of normal prion produces a particular pathology when it spontaneously deforms to produce sCJD.
But if these proteins deform because of infection with a disease-causing prion, the relationship between pathology and prion type should be different, as it is in vCJD. "If we look at brain samples from sporadic CJD cases and find some that do not fit the pattern," says Kretschmar, "that could mean they were caused by infection."
There are 250 deaths per year from sCJD in the US, and a similar incidence elsewhere. Singeltary and other US activists think that some of these people died after eating contaminated meat or "nutritional" pills containing dried animal brain. Governments will have a hard time facing activists like Singeltary if it turns out that some sCJD isn't as spontaneous as doctors have insisted.
Deslys's work on macaques also provides further proof that the human disease vCJD is caused by BSE. And the experiments showed that vCJD is much more virulent to primates than BSE, even when injected into the bloodstream rather than the brain. This, says Deslys, means that there is an even bigger risk than we thought that vCJD can be passed from one patient to another through contaminated blood transfusions and surgical instruments.
REPORT OF THE ADVISORY COMMITTEE ON SCRAPIE 1976
IN CONFIDENCE
Such considerations suggest first that those responsible for work with scrapie should be selected with as much care as are astronauts.
THURSDAY, NOVEMBER 9, 2023
EFSA Annual Report of the Scientific Network on BSE-TSE 2023
Annual Report of the Scientific Network on BSE-TSE 2023
European Food Safety Authority (EFSA
APPROVED: 25 October 2023
Monday, November 13, 2023
Food and Drug Administration's BSE Feed Regulation (21 CFR 589.2000) Singeltary Another Request for Update 2023
Unforeseen decrease of full-length prion protein in macaques exposed to prion contaminated blood products
Emmanuel COMOY, Nina JAFFRE, Jérôme DELMOTTE, Jacqueline MIKOL, and Jean Philippe DESLYS Commissariat à l’Energie Atomique, DRF/IBFJ/SEPIA, 18 Route du Panorama, 92265 Fontenay-aux-Roses, France.
Aims: The presence of prion infectivity in blood from patients affected by variant of Creutzfeldt-Jakob disease (v-CJD) questions the risk of its inter-human transmission through transfusion. We have previously described that several cynomolgus macaques experimentally exposed to prion-contaminated blood products developed c-BSE/v-CJD; however, after an exposure to low infectious doses, the vast majority of them developed an unexpected, fatal disease phenotype focused on spinal cord involvement which does not fulfill the classical diagnostic criteria of v-CJD, notably concerning the pathognomonic accumulation of abnormal prion protein. Here we aim to investigate the etiology and physiopathology of this original myelopathy.
Materials and Methods: CNS (brain and spinal cord) samples from myelopathic macaques were tested with different biochemical approaches in comparison to samples derived from either healthy animals or their counterparts exposed to different strains of prion diseases.
Results: Current conventional techniques failed to detect any accumulation of abnormal prion protein (PrPv-CJD) in the CNS of these myelopathic animals. Conversely, in their spinal cord we observed an alteration of their physiological cellular PrP pattern: PrP was not detectable under its full-length classical expression but mainly under its physiological terminal-truncated C1 fragment.
Conclusions: We here confirm the prion origin of this original syndrome, with a very specific biochemical signature linked to changes in PrP at the level of spinal cord lesions: contrary to what is classically described in prion diseases, host PrP is here altered in a form that is abnormally sensitive to degradation by cellular catabolism. This could provide the first experimental evidence of a link between loss of function of the cellular prion protein and the onset of disease. These observations open up new horizons in the field of prion diseases, which has hitherto been limited to pathologies associated with abnormal changes in cellular PrP towards highly structured conformations, with the possibility of unsuspected prion mechanisms/origins in certain neurodegenerative disorders.
Funded by: Financial support for the study was provided by the French National Research Agency (ANR).
Grant number: ANR-10-BLAN-133001 and BIOTECS2010-BloodSecur
Acknowledgement: We specially thank N. Lescoutra, A. Culeux, V. Durand, E. Correia, C. Durand and S. Jacquin for precious technical assistance
Transmission of prion infectivity from CWD-infected macaque tissues to rodent models demonstrates the zoonotic potential of chronic wasting disease
Samia Hannaoui1,2, Ginny Cheng1,2, Wiebke Wemheuer3, Walter Schulz-Schaeffer3, Sabine Gilch1,2, Hermann Schatzl1,2 1University of Calgary, Calgary, Canada. 2Calgary Prion Research Unit, Calgary, Canada. 3Institute of Neuropathology, Medical Faculty, Saarland University, Homburg/Saar, Germany
***> Further passage to cervidized mice revealed transmission with a 100% attack rate.
***> Our findings demonstrate that macaques, considered the best model for the zoonotic potential of prions, were infected upon CWD challenge, including the oral one.
****> The disease manifested as atypical in macaques and initial transgenic mouse transmissions, but with infectivity present at all times, as unveiled in the bank vole model with an unusual tissue tropism.
***> Epidemiologic surveillance of prion disease among cervid hunters and people likely to have consumed venison contaminated with chronic wasting disease
=====
Transmission of Cervid Prions to Humanized Mice Demonstrates the Zoonotic Potential of CWD
Samia Hannaouia, Irina Zemlyankinaa, Sheng Chun Changa, Maria Immaculata Arifina, Vincent Béringueb, Debbie McKenziec, Hermann M. Schatzla, and Sabine Gilcha
Results: Here, we provide the strongest evidence supporting the zoonotic potential of CWD prions, and their possible phenotype in humans. Inoculation of mice expressing human PrPCwith deer CWD isolates (strains Wisc-1 and 116AG) resulted in atypical clinical manifestations in > 75% of the mice, with myoclonus as leading clinical sign. Most of tg650brain homogenates were positive for seeding activity in RT-QuIC. Clinical disease and presentation was transmissible to tg650 mice and bank voles. Intriguingly, protease-resistant PrP in the brain of tg650 mice resembled that found in a familial human prion disease and was transmissible upon passage. Abnormal PrP aggregates upon infection with Wisc-1 were detectable in thalamus, hypothalamus, and midbrain/pons regions.
Unprecedented in human prion disease, feces of CWD-inoculated tg650 mice harbored prion seeding activity and infectious prions, as shown by inoculation of bank voles and tg650 with fecal homogenates.
Conclusions: This is the first evidence that CWD can infect humans and cause disease with a distinctive clinical presentation, signature, and tropism, which might be transmissible between humans while current diagnostic assays might fail to detect it. These findings have major implications for public health and CWD-management.
The finding that infectious PrPSc was shed in fecal material of CWD-infected humanized mice and induced clinical disease, different tropism, and typical three banding pattern-PrPres in bank voles that is transmissible upon second passage is highly concerning for public health. The fact that this biochemical signature in bank voles resembles that of the Wisc-1 original deer isolate and is different from that of bvWisc-1, in the migration profile and the glyco-form-ratio, is valid evidence that these results are not a product of contamination in our study. If CWD in humans is found to be contagious and transmissible among humans, as it is in cervids [57], the spread of the disease within humans might become endemic.
Transmission of cervid prions to humanized mice demonstrates the zoonotic potential of CWD
Acta Neuropathol 144, 767–784 (2022). https://doi.org/10.1007/s00401-022-02482-9
Published
22 August 2022
Transmission of cervid prions to humanized mice demonstrates the zoonotic potential of CWD
Samia Hannaoui1 · Irina Zemlyankina1 · Sheng Chun Chang1 · Maria Immaculata Arifn1 · Vincent Béringue2 · Debbie McKenzie3 · Hermann M. Schatzl1 · Sabine Gilch1
Accepted: 7 August 2022
HIGHLIGHTS OF THIS STUDY
Our results suggest that CWD might infect humans, although the transmission barrier is likely higher compared to zoonotic transmission of cattle prions. Notably, our data suggest a different clinical presentation, prion signature, and tissue tropism, which causes challenges for detection by current diagnostic assays. Furthermore, the presence of infectious prions in feces is concerning because if this occurs in humans, it is a source for human-to-human transmission. These findings have strong implications for public health and CWD management.
Our results are the first evidence of a zoonotic risk of CWD when using one of the most common CWD strains, Wisc-1/CWD1 for infection. We demonstrated in a human transgenic mouse model that the species barrier for transmission of CWD to humans is not absolute.
Our findings strongly suggest that CWD should be regarded as an actual public health risk. Here, we use humanized mice to show that CWD prions can cross the species barrier to humans, and remarkably, infectious prions can be excreted in feces.
suggesting a potential for human-to-human transmission and a real iatrogenic risk that might be unrecognizable.
If CWD in humans is found to be contagious and transmissible among humans, as it is in cervids [57], the spread of the disease within humans might become endemic.
Supplementary Information The online version contains supplementary material available at
snip...see full text;
Detection of chronic wasting disease prions in processed meats
Rebeca Benavente1 , Francisca Bravo1,2, J. Hunter Reed3 , Mitch Lockwood3 , Glenn Telling4 , Rodrigo Morales1,2 1 Department of Neurology, McGovern Medical School, University of Texas Health Science Center at Houston, Texas, USA; 2 Universidad Bernardo O’Higgins. Santiago, Chile; 3 Texas Parks and Wildlife Department, Texas, USA. 4 Prion Research Center, Department of Microbiology, Immunology, and Pathology, Colorado State University, Fort Collins, CO, USA
Aims: identify the presence of CWD prions in processed meats derived from elk.
Materials and Methods: In this study, we analyzed different processed meats derived from a CWD-positive (pre-clinical) free-ranging elk. Products tested included filets, sausages, boneless steaks, burgers, seasoned chili meats, and spiced meats. The presence of CWD-prions in these samples were assessed by PMCA using deer and elk substrates. The same analyses were performed in grilled and boiled meats to evaluate the resistance of the infectious agent to these procedures.
Results: Our results show positive prion detection in all the samples analyzed using deer and elk substrates. Surprisingly, cooked meats displayed increased seeding activities. This data suggests that CWD-prions are available to people even after meats are processed and cooked.
Conclusions: These results suggest CWD prions are accessible to humans through meats, even after processing and cooking. Considering the fact that these samples were collected from already processed specimens, the availability of CWD prions to humans is probably underestimated.
Funded by: NIH and USDA
Grant number: 1R01AI132695 and APP-20115 to RM
Acknowledgement: We would like to thank TPWD personnel for providing us with valuable samples
"Our results show positive prion detection in all the samples analyzed using deer and elk substrates. Surprisingly, cooked meats displayed increased seeding activities."
end...
PRION 2023 CONTINUED;
Fortuitous generation of a zoonotic cervid prion strain
Manuel Camacho, Xu Qi, Liuting Qing, Sydney Smith, Jieji Hu, Wanyun Tao, Ignazio Cali, Qingzhong Kong. Department of Pathology, Case Western Reserve University, Cleveland, USA
Aims: Whether CWD prions can infect humans remains unclear despite the very substantial scale and long history of human exposure of CWD in many states or provinces of USA and Canada. Multiple in vitro conversion experiments and in vivo animal studies indicate that the CWD-to-human transmission barrier is not unbreakable. A major long-term public health concern on CWD zoonosis is the emergence of highly zoonotic CWD strains. We aim to address the question of whether highly zoonotic CWD strains are possible.
Materials and Methods: We inoculated several sCJD brain samples into cervidized transgenic mice (Tg12), which were intended as negative controls for bioassays of brain tissues from sCJD cases who had potentially been exposed to CWD. Some of the Tg12 mice became infected and their brain tissues were further examined by Western blot as well as serial passages in humanized or cervidized mice.
Results: Passage of sCJDMM1 in transgenic mice expressing elk PrP (Tg12) resulted in a “cervidized” CJD strain that we termed CJDElkPrP. We observed 100% transmission of the original CJDElkPrP in transgenic mice expressing human PrP. We passaged CJDElkPrP two more times in the Tg12 mice. We found that such second and third passage CJDElkPrP prions retained 100% transmission rate in the humanized mice, despite that the natural elk CWD isolates and CJDElkPrP share the same elk PrP sequence. In contrast, we and others found zero or poor transmission of natural elk CWD isolates in humanized mice.
Conclusions: Our data indicate that highly zoonotic cervid prion strains are not only possible but also can retain zoonotic potential after serial passages in cervids, suggesting a very significant and serious long-term risk of CWD zoonosis given that the broad and continuing spread of CWD prions will provide fertile grounds for the emergence of zoonotic CWD strains over time.
Funded by: NIH Grant number: R01NS052319, R01NS088604, R01NS109532
Acknowledgement: We want to thank the National Prion Disease Pathology Surveillance Center and Drs. Allen Jenny and Katherine O'Rourke for providing the sCJD samples and the CWD samples used in this study, respectively
"Our data indicate that highly zoonotic cervid prion strains are not only possible but also can retain zoonotic potential after serial passages in cervids, suggesting a very significant and serious long-term risk of CWD zoonosis given that the broad and continuing spread of CWD prions will provide fertile grounds for the emergence of zoonotic CWD strains over time."
PRION 2023 CONTINUED;
A probable diagnostic marker for CWD infection in humans
Xu Qi, Liuting Qing, Manuel Camacho, Ignazio Cali, Qingzhong Kong. Department of Pathology, Case Western Reserve University, Cleveland, USA
Aims: Multiple in vitro CWD-seeded human PrP conversion experiments and some animal model studies indicate that the species barrier for CWD to human transmission can be overcome, but whether CWD prion can infect humans in real life remains controversial. The very limited understanding on the likely features of CWD infection in humans and the lack of a reliable diagnostic marker for identification of acquired human CWD cases contribute to this uncertainty. We aim to stablish such a reliable diagnostic marker for CWD infections in humans should they occur.
Materials and Methods: A couple of PrPSc-positive spleens were identified from humanized transgenic mice inoculated with either CWD or sCJDMM1. Prions in these spleens were compared by bioassays in cervidized or humanized transgenic mice. A couple of PrPSc-positive spleens from UK sCJDMM1 patients were also examined similarly as controls with no exposure to CWD.
Results: We have detected two prion-positive spleens in humanized transgenic mice inoculated with some CWD isolates. Such experimentally generated splenic “humanized” CWD prions (termed eHuCWDsp) appear indistinguishable from prions in the brain of sCJDMM1 patients on Western blot. We compared eHuCWDsp with prions in the spleen from humanized mice infected with sCJDMM1 (termed sCJDMM1sp) by bioassays in cervidized or humanized transgenic mice. Significantly, we found that eHuCWDsp can efficiently infect not only the humanized mice but also cervidized transgenic mice, and cervidized mice infected by eHuCWDsp produced PrPSc and brain pathology that are practically identical to those of CWD-infected cervidized mice. In contrast, sCJDMM1sp, similar to prions from sCJDMM1 patient brains, is poorly transmissible in the cervidized mice.
Conclusions: Our data demonstrate that high transmissibility with CWD features of splenic prions in cervidized transgenic mice is unique to acquired human CWD prions, and it may serve as a reliable marker to identify the first acquired human CWD cases.
Funded by: NIH Grant number: R01NS052319, R01NS088604, R01NS109532
Acknowledgement: We want to thank the National Prion Disease Pathology Surveillance Center and Drs. Allen Jenny and Katherine O'Rourke for providing the sCJD samples and the CWD samples used in this study, respectively.
=====end
PRION 2023 CONTINUED;
Prion 2023 Experimental Oronasal Inoculation of the Chronic Wasting Disease Agent into White Tailed Deer
Author list: Sarah Zurbuchena,b , S. Jo Moorea,b , Jifeng Biana , Eric D. Cassmanna , and Justin J. Greenleea . a. Virus and Prion Research Unit, National Animal Disease Center, ARS, United States Department of Agriculture, Ames, IA, US b. Oak Ridge Institute for Science and Education (ORISE), U.S. Department of Energy, Oak Ridge, TN, United States
Aims: The purpose of this experiment was to determine whether white-tailed deer (WTD) are susceptible to inoculation of chronic wasting disease (CWD) via oronasal exposure.
Materials and methods: Six male, neutered WTD were oronasally inoculated with brainstem material (10% w/v) from a CWD-positive wild-type WTD. The genotypes of five inoculated deer were Q95/G96 (wild-type). One inoculated deer was homozygous S at codon 96 (96SS). Cervidized (Tg12; M132 elk PrP) mice were inoculated with 1% w/v brainstem homogenate from either a 96GG WTD (n=10) or the 96SS WTD (n=10).
Results: All deer developed characteristic clinical signs of CWD including weight loss, regurgitation, and ataxia. The 96SS individual had a prolonged disease course and incubation period compared to the other deer. Western blots of the brainstem on all deer yielded similar molecular profiles. All deer had widespread lymphoid distribution of PrPCWD and neuropathologic lesions associated with transmissible spongiform encephalopathies. Both groups of mice had a 100% attack rate and developed clinical signs, including loss of body condition, ataxia, and loss of righting reflex. Mice inoculated with material from the 96SS deer had a significantly shorter incubation period than mice inoculated with material from 96GG deer (Welch two sample T-test, P<0.05). Serial dilutions of each inocula suggests that differences in incubation period were not due to a greater concentration of PrPCWD in the 96SS inoculum. Molecular profiles from western blot of brain homogenates from mice appeared similar regardless of inoculum and appear similar to those of deer used for inoculum.
Conclusions: This study characterizes the lesions and clinical course of CWD in WTD inoculated in a similar manner to natural conditions. It supports previous findings that 96SS deer have a prolonged disease course. Further, it describes a first pass of inoculum from a 96SS deer in cervidized mice which shortened the incubation period.
Funded by: This research was funded in its entirety by congressionally appropriated funds to the United States Department of Agriculture, Agricultural Research Service. The funders of the work did not influence study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Acknowledgement: We thank Ami Frank and Kevin Hassall for their technical contributions to this project.
=====end
PRION 2023 CONTINUED;
''Currently, there is scientific evidence to suggest that CWD has zoonotic potential; however, no confirmed cases of CWD have been found in humans.''
PART 2. TPWD CHAPTER 65. DIVISION 1. CWD
31 TAC §§65.82, 65.85, 65.88
The Texas Parks and Wildlife Commission in a duly noticed meeting on May 25, 2023 adopted amendments to 31 TAC §§65.82, 65.85, and §65.88, concerning Disease Detection and Response, without changes to the proposed text as published in the April 21, 2023, issue of the Texas Register (48 TexReg 2048). The rules will not be republished.
Currently, there is scientific evidence to suggest that CWD has zoonotic potential; however, no confirmed cases of CWD have been found in humans.
17 DETECTION OF CHRONIC WASTING DISEASE PRIONS IN PROCESSED MEATS.
Rebeca Benavente1, Francisca Bravo1,2, Paulina Soto1,2, J. Hunter Reed3, Mitch Lockwood3, Rodrigo Morales1,2
1Department of Neurology, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, USA. 2Universidad Bernardo O’Higgins, Santiago, Chile. 3Texas Parks and Wildlife, Austin, USA
Abstract
The zoonotic potential of chronic wasting disease (CWD) remains unknown. Currently, there are no known natural cases of CWD transmission to humans but increasing evidence suggests that the host range of CWD is not confined only to cervid species. Alarmingly, recent experimental evidence suggests that certain CWD isolates can induce disease in non-human primates. While the CDC strongly recommends determining CWD status in animals prior to consumption, this practice is voluntary. Consequently, it is plausible that a proportion of the cervid meat entering the human food chain may be contaminated with CWD. Of additional concern is that traditional diagnostic techniques used to detect CWD have relatively low sensitivity and are only approved for use in tissues other than those typically ingested by humans. In this study, we analyzed different processed meats derived from a pre-clinical, CWD-positive free-ranging elk. Products tested included filets, sausages, boneless steaks, burgers, ham steaks, seasoned chili meats, and spiced meats. CWD-prion presence in these products were assessed by PMCA using deer and elk substrates. Our results show positive prion detection in all products. To confirm the resilience of CWD-prions to traditional cooking methods, we grilled and boiled the meat products and evaluated them for any remnant PMCA seeding activity. Results confirmed the presence of CWD-prions in these meat products suggesting that infectious particles may still be available to people even after cooking. Our results strongly suggest ongoing human exposure to CWD-prions and raise significant concerns of zoonotic transmission through ingestion of CWD contaminated meat products.
***> Products tested included filets, sausages, boneless steaks, burgers, ham steaks, seasoned chili meats, and spiced meats.
***> CWD-prion presence in these products were assessed by PMCA using deer and elk substrates.
***> Our results show positive prion detection in all products.
***> Results confirmed the presence of CWD-prions in these meat products suggesting that infectious particles may still be available to people even after cooking.
***> Our results strongly suggest ongoing human exposure to CWD-prions and raise significant concerns of zoonotic transmission through ingestion of CWD contaminated meat products.
=====
9 Carrot plants as potential vectors for CWD transmission.
Paulina Soto1,2, Francisca Bravo-Risi1,2, Claudio Soto1, Rodrigo Morales1,2
1Department of Neurology, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, USA. 2Universidad Bernardo O’Higgins, Santiago, Chile
***> We show that edible plant components can absorb prions from CWD-contaminated soils and transport them to their aerial parts.
***> Our results indicate that edible plants could participate as vectors of CWD transmission.
=====
Transmission of prion infectivity from CWD-infected macaque tissues to rodent models demonstrates the zoonotic potential of chronic wasting disease.
Samia Hannaoui1,2, Ginny Cheng1,2, Wiebke Wemheuer3, Walter Schulz-Schaeffer3, Sabine Gilch1,2, Hermann Schatzl1,2 1University of Calgary, Calgary, Canada. 2Calgary Prion Research Unit, Calgary, Canada. 3Institute of Neuropathology, Medical Faculty, Saarland University, Homburg/Saar, Germany
***> Further passage to cervidized mice revealed transmission with a 100% attack rate.
***> Our findings demonstrate that macaques, considered the best model for the zoonotic potential of prions, were infected upon CWD challenge, including the oral one.
****> The disease manifested as atypical in macaques and initial transgenic mouse transmissions, but with infectivity present at all times, as unveiled in the bank vole model with an unusual tissue tropism.
***> Epidemiologic surveillance of prion disease among cervid hunters and people likely to have consumed venison contaminated with chronic wasting disease
=====
Transmission of Cervid Prions to Humanized Mice Demonstrates the Zoonotic Potential of CWD
Samia Hannaouia, Irina Zemlyankinaa, Sheng Chun Changa, Maria Immaculata Arifina, Vincent Béringueb, Debbie McKenziec, Hermann M. Schatzla, and Sabine Gilcha
Results: Here, we provide the strongest evidence supporting the zoonotic potential of CWD prions, and their possible phenotype in humans. Inoculation of mice expressing human PrPCwith deer CWD isolates (strains Wisc-1 and 116AG) resulted in atypical clinical manifestations in > 75% of the mice, with myoclonus as leading clinical sign. Most of tg650brain homogenates were positive for seeding activity in RT-QuIC. Clinical disease and presentation was transmissible to tg650 mice and bank voles. Intriguingly, protease-resistant PrP in the brain of tg650 mice resembled that found in a familial human prion disease and was transmissible upon passage. Abnormal PrP aggregates upon infection with Wisc-1 were detectable in thalamus, hypothalamus, and midbrain/pons regions.
Unprecedented in human prion disease, feces of CWD-inoculated tg650 mice harbored prion seeding activity and infectious prions, as shown by inoculation of bank voles and tg650 with fecal homogenates.
Conclusions: This is the first evidence that CWD can infect humans and cause disease with a distinctive clinical presentation, signature, and tropism, which might be transmissible between humans while current diagnostic assays might fail to detect it. These findings have major implications for public health and CWD-management.
WEDNESDAY, NOVEMBER 01, 2023
TEXAS CHRONIC WASTING DISEASE RISES SUBSTANTIALLY TO 575 CONFIRMED CWD CASES TO DATE
FRIDAY, DECEMBER 08, 2023
TEXAS CWD TSE PRION DIRE CONSEQUENCES ARE HERE!
THURSDAY, DECEMBER 7, 2023
Chronic Wasting Disease CWD TSE Prion Cervid Update By State December 2023
(Long Version)
(Short Version)
NOW, THINK CONSUMPTION, EXPOSURE, FRIENDLY FIRE, PASS IT FORWARD, IATROGENIC CJD!
Friday, October 20, 2023
An investigation has been opened into the death of a scientist who was studying a transmissible and deadly disease CJD in Spain DEGENERATIVE DISEASES
An investigation has been opened into the death of a scientist who was studying a transmissible and deadly disease in Spain
Three institutions are trying to ascertain the origin of the infectious Creutzfeldt-Jakob disease samples discovered in the biochemist’s laboratory. The 45-year-old investigator died in 2022
The University of Barcelona’s School of Medicine, in L’Hospitalet de Llobregat, where laboratory 4141 is located.
MASSILIANO MINOCRI
Manuel Ansede
MANUEL ANSEDE
Madrid - OCT 19, 2023 - 16:15 EDT
A prestigious Spanish researcher of Creutzfeldt-Jakob disease died last year after experiencing symptoms consistent with this deadly ailment, as EL PAÍS has learned from multiple sources at the three institutions involved. Three months ago, the University of Barcelona opened an internal investigation to ascertain the origin of thousands of unauthorized samples, some of them infectious, discovered in a freezer in its laboratory 4141, where the deceased biochemist worked. He was a member of the Bellvitge Biomedical Research Institute (IDIBELL) and the CIBER public consortium. These two institutions have joined the internal investigation, after noting concern among colleagues at the facility, who did not know the level of risk to which they were exposed without their knowledge. This neurodegenerative disease incubates silently for years, but when symptoms appear — rapid dementia and muscle stiffness — it is fatal. Life expectancy after diagnosis is barely six months. Its best-known animal equivalent is mad cow disease.
The biochemist joined the 4141 lab at the University of Barcelona in January 2018 as a principal investigator with a group of his own; his wife joined shortly after. Together, they identified characteristic substances in human cerebrospinal fluid, useful for the diagnosis of rapid dementia. In November 2020, the now deceased scientist began to feel unwell and asked to leave. After his colleagues found out that his symptoms were consistent with Creutzfeldt-Jakob disease, he demanded absolute privacy and decided to hide his diagnosis, according to the sources consulted for this article. He died at the age of 45.
On December 18, 2020, the head of the 4141 laboratory, Isidre Ferrer, a professor of Pathology at the University of Barcelona and a member of IDIBELL, informed the directors of both institutions that suspicious samples of cerebrospinal fluid from people with Creutzfeldt-Jakob disease and other neurodegenerative types of dementia had been discovered by chance in a freezer at 80 degrees below zero, according to internal documentation to which EL PAÍS had access. The thousands of unauthorized samples from patients and animals were in a drawer reserved for the sick researcher’s group and lacked records indicating their presence. The University of Barcelona then ordered the immediate closure and decontamination of laboratory 4141, located in the School of Medicine at L’Hospitalet de Llobregat.
Doctor Gabriel Capellá, the director of IDIBELL, explains that they have identified “a maximum of eight people” who worked in the laboratory at that time, in addition to the deceased scientist and Isidre Ferrer. Some of these coworkers have required months of psychological care. The university’s safety office and IDIBELL’s prevention service determined that there was “an unacceptable risk,” although Capellá emphasizes that “there is no record of any occupational accident” in which a researcher could have been infected with contaminated material. Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies are caused by abnormal proteins called prions, which accumulate in the brain and cause a microscopic sponge-like appearance. There are only one or two cases per million inhabitants, the vast majority of which are of unknown cause, but cases of the disease have also been reported after contact with surgical instruments contaminated by these prions.
The three institutions involved took more than two years to send the suspect samples for analysis to a specialized center, the CIC bioGUNE, in Derio, Spain. A spokeswoman for the University of Barcelona says that they sent them in December 2022 and the three organizations received the results in March 2023. Four months later, in July, the legal departments at the three institutions finally informed the 4141 laboratory workers that the Creutzfeldt-Jakob disease samples were potentially infectious, as feared. “You can debate whether we have been quick [in our response] or not, but we have been transparent. We are [part of] three institutions that had to agree, and we have acted as guarantors,” says Capellá. A similar situation also occurred in France; following the death of a researcher from Creutzfeldt-Jakob in 2019 and the discovery of another suspected case, all public laboratories investigating prion diseases decided to temporarily close in July 2021 to review their protocols.
Laboratory 4141 was not equipped to handle high biohazard samples. It did not even have a biosafety hood. At the end of 2018, the CIBER public consortium signed an agreement so that the group could work with these dangerous samples at the high-security laboratory of the Animal Health Research Center (CReSA) in Bellaterra, Spain, near Barcelona. According to the sources we consulted, there was no reason to have the contaminated material in laboratory 4141, beyond saving time during experiments, since the CReSA bunker is 30 kilometers (about 19 miles) away and required waiting one’s turn to use. Isidre Ferrer, the head of the facility at the time, who has since retired, prefers not to comment on the case until the internal investigation is completed, but he emphasizes that he was unaware of the existence of these dangerous samples.
The IDIBELL director recalls that the deceased scientist was “a promising and brilliant researcher.” From 2013 to 2017, he worked at the University Medical Center of Göttingen (Germany) under neurologist Inga Zerr, a leading international expert in Creutzfeldt-Jakob disease. Physician Margarita Blázquez, who manages the CIBER public consortium, notes that the disease’s incubation period can last several years, so, if the deceased researcher really had it, he also could have become infected with it in Germany or at another of his previous laboratories. This newspaper has tried to contact the scientist’s widow via email but has not received a response. She asked to be discharged shortly after her husband did. The three institutions are now investigating whether the couple handled the dangerous samples without authorization in lab 4141. A third person affiliated with CIBER, a member of the now-deceased biochemist’s research group, worked with potentially infectious Creutzfeldt-Jakob samples without being informed that they were infectious.
The security office of the University of Barcelona believes that the samples would only have been a problem in the case of accidental inoculation or ingestion while handling them. But internal documents confirm the alarm the situation has caused on campus. “The laboratory technicians and investigators express their enormous concern about the fact that, so far, it has not been possible to determine the origin of the doctor’s illness. They are left to worry about whether they may suffer the same fate in a few years’ time as a result of uncontrolled contamination that may have been created in the laboratory,” according to the minutes of a December 22, 2020, meeting between workers and Carles Solsona, the director of the Department of Pathology at the University of Barcelona. “This fear causes them to suffer a state of permanent anguish, causing insomnia and irritability.”
The IDIBELL director sent a message to the center’s entire staff on the 11th, five days after EL PAÍS informed him that it was investigating the case. Gabriel Capellá then told his workers of “a very serious incident that became known on campus for the first time at the end of 2020.″ With “deep dismay,” Capellá announced the researcher’s death “due to a possible prion condition,” with “a possible iatrogenic [a disease acquired by contact with contaminated materials during a medical procedure].” The director also reported finding “potentially dangerous samples” in a freezer. “Our priority is to ensure that this situation is handled rigorously and transparently to limit the damage to the reputation of our institutions,” he said.
Do you have more information about this case or other similar ones? You can write to us at mansede@elpais.es.
Sign up for our weekly newsletter to get more English-language news coverage from EL PAÍS USA Edition
Friendly fire, pass it forward, they call it iatrogenic cjd, or what i call 'tse prion poker', are you all in $$$
all iatrogenic cjd is, is sporadic cjd, before the iatrogenic event is discovered, traced back, proven, documented, put into the academic domain, and then finally the public domain, this very seldom happens, thus problem solved, it's all sporadic cjd...
Direct neural transmission of vCJD/BSE in macaque after finger incision CORRESPONDENCE
Direct neural transmission of vCJD/BSE in macaque after finger incision
Jacqueline Mikol1 · Jérôme Delmotte1 · Dolorès Jouy1 · Elodie Vaysset1 · Charmaine Bastian1 · Jean‑Philippe Deslys1 ·
Emmanuel Comoy1 Received: 10 July 2020 / Revised: 8 September 2020 / Accepted: 25 September 2020 / Published online: 6 October 2020 © The Author(s) 2020
Non-human primates appeared as the closest model to study human iatrogenic prion diseases [14]: we report here the consequences of variant Creutzfeldt–Jakob disease/bovine spongiform encephalopathy (vCJD/BSE) inoculation in a cynomolgus macaque finger, with the demonstration of an original mode of propagation and the practical risk for professional exposure.
The distal right middle finger handpad of a 4-year-old macaque was incised on both lateral sides to induce local inflammation, and then injected with the equivalent of 10 mg of a BSE, orally challenged macaque brain [18]. After an 18 months period of finger clumsiness, the clinical disease (behaviour abnormalities, fear, hyperesthesia, gait disturbances, shaking) began 7.5 years after inoculation and euthanasia took place 2 months later for welfare reasons. Motor conduction velocity of the right median nerve was reduced to one-third of the left counterpart and sensory potential was not detected.
Histological and biochemical studies were performed as previously described. All the elements of the triad were present [7–9]: spongiform change was moderate in neocortex, striatum, brain stem, mild in spinal cord but severe in thalamus and cerebellum; neuronal loss was globally moderate, but severe in cerebellum and sacral spinal cord (vacuolated neurons); gliosis was severe in thalamus, cerebellum and brain stem and moderate elsewhere (Supplementary Fig. 1). ELISA and western blot (WB) showed the expected accumulation of PrPres with BSE glycophoretic pattern at all levels of brain and spinal cord (Supplementary Fig. 2).
In the brain, PrPd deposits were laminar into the cortical deep layers, massive into thalamus, basal ganglia, cerebellum, and brain stem. In spinal cord, PrPd was symmetrically distributed, intense in the Substantia gelatinosa and nucleus dorsal of Clarke while decreased at sacral level. Deposits were diverse into the whole CNS: synaptic, perineuronal, reticular aggregates, mini-plaques, plaques, and incomplete florid plaques. The retinal plexiform layers were labelled (Supplementary Fig. 1i). There were no amyloid or tau deposits.
Unusual PrPd deposits were observed along dendrites, short and long axons, neuritic threads tracing fne networks of straight lines or like strings of pearls (Supplementary Fig. 3). They were present into deep neocortex, basal ganglia, and motoneurons. Such long processes are not frequent but have been reported in human [13] and experimental studies [10, 22]. PrPd deposits were also noted as very mild into striato-pallidal projections, both limbs of internal capsule and fornix (Supplementary Fig. 3). The presence of PrPd in white matter has been reported (Supplementary text 4).
Peripherally, the expected PrPd was undetectable in lymphoid organs, including spleen, through biochemical or immunohistochemical analyses, while prion replication was detected in the peripheral nervous system (PNS): PrPd staining was visualized in many dorsal root ganglia (DRG) but only in nerves innervating the forelimb site of injection (median and ulnar nerves). At the cellular level, PrPd was limited to ganglia and satellite cells in DRG and Schwann cells (Scs) all along nerves whereas axons were never labelled (Fig. 1). Previously, using postmortem immunohistochemical studies (listed in Supplementary text 5), PrPd has been shown in peripheral nervous system in all forms of human neuropathies, albeit more frequently in vCJD, mostly in posterior root nerve fbres at adaxonal location and/or in ganglion and satellite cells. The restricted amount of PrPd was repeatedly underlined but, recently, prion RTQuiC was positive in all nerves examined [2]. PrPd has also been described, frst in scrapie [17] then in BSE, as limited “adaxonal deposits” or/and Sc deposits, with or without DRG cell involvement (review in [4] and Supplementary text 6). Previous studies of the mode of propagation of PrPd have reported variable observations and analyses depending on strains, host species and genotype (Supplementary text 6); the authors discussed the role of the sensory route of trafficking of prions, the modifications of axonal transport, the centrifugal versus centripetal spread of PrPd .
After peripheral infection, accumulation of infectious agent is reputed to occur in lymphoid tissues before direct neuroinvasion [18, 19], even with very little apparent peripheral lymphoreticular deposition [6, 20]. Here, there is no apparent replication/amplification of vCJD/BSE agent in the lymphoid tissues of the exposed macaque. In this model, the neural contamination occurred directly in the highly innervated finger while neuroinvasion appears to occur in Scs along the median nerve to the DRG, with the appearance of the classical labelling of ganglion cells which indicates the onset of the first level of neuronal infection. This model provides direct evidence of the hypothesis of a sequential infection of Scs from the periphery to the CNS, followed by a secondary diffusion into the spinal cord, as already considered by our group [15] and others [1, 3, 11, 12, 21]. It is to note that studies based on intra-sciatic nerve injections in hamsters [16] and transgenic mice [12] had established a rate of transport of infectivity of, respectively, 0.5–2 mm and 0.7 mm per day. This key role of Scs could explain both the low speed of propagation and the discrepancy between the paucity of PrPd into the distal part of the sensory nerves followed by the positivity of DRG, satellite cells and proximal roots.
In conclusion, we have observed that the exposure of a primate to vCJD/BSE through a distal finger lesion induces, after more than 7.5 years of silent incubation, a massive deposit of PrPd , strictly restricted to the nervous system and the eye.
Our data suggest a new type of pure unique peripheral nervous contamination in which the Scs would have a major role in the mode of centripetal progression of PrPd in the peripheral nervous system. Moreover, considering the fact that, recently, “a variant CJD diagnosed 7.5 years after occupational exposure” (cryomicrotomy) in a technician was observed [5], this experimental case report supports the risk linked to professional exposure and reinforces the necessity of adequate measures of prevention.
Second death in France in a laboratory working on prions
Creutzfeldt-Jakob disease has killed a person who handled this infectious agent at Inrae in Toulouse. After a first death in 2019, a moratorium on work on this pathogen has been extended.
By Hervé Morin
Creutzfeldt-Jakob disease killed a few days ago a retired research technician from the National Research Institute for Agriculture, Food and the Environment (Inrae), who had worked in Toulouse in contact of biological tissue infected with prions. This death sows consternation and concern in the scientific community working with these infectious agents. It follows the death, on June 17, 2019, of Emilie Jaumain, a 33-year-old laboratory technician, suffering from the same incurable neurodegenerative disease. The young woman is said to have contracted it in 2010, cutting herself while handling fragments of the brains of mice infected with prions, in another unit of INRAE, in Jouy-en-Josas.
Computer representation of part of a prion protein on a light micrograph of pyramidal nerve cells (neurons, in black) in the cerebellum of the brain. ALFRED PASIEKA / SCIENCE PHOTO LIBRARY
Regarding the retiree from Toulouse, it will be necessary to determine whether she was the victim of a genetic or sporadic form of Creutzfeldt-Jakob disease, if the disease may have been caused by the ingestion of meat contaminated by the agent of encephalopathy. bovine spongiform (BSE, also called mad cow disease) or, as in the case of Emilie Jaumain, if accidental occupational exposure can be claimed. Prion diseases are caused by proteins taking an aberrant conformation, which gives them the property of replicating to form aggregates that are deleterious for neurons. There are around 150 cases per year in France, resulting in fatal degeneration of the central nervous system.
Temporary suspension of work on prions in French public research laboratories
PRESS RELEASE - The general directorates of ANSES, CEA, CNRS, INRAE and Inserm, have decided jointly and in agreement with the Ministry of Higher Education, Research and Innovation to suspend as a precaution all their research and experimentation work relating to prion diseases, for a period of three months.
This precautionary measure is motivated by the knowledge of a possible new case of a person suffering from Creutzfeldt-Jakob disease and who worked in a laboratory for research on prions.
Posted on July 27, 2021
The suspension period put in place as of this day will make it possible to study the possibility of a link between the observed case and the person's former professional activity and to adapt, if necessary, the preventive measures in force in the research laboratories.
The person with Creutzfeldt-Jakob disease (CJD)1, whose form is not yet known, is a retired INRAE agent. This could be the second case of infectious CJD affecting a scientist who worked on prions, after that of an assistant engineer who died of the disease in 2019, and who was injured in 2010 during of an experiment.
Following this death, a general inspection mission was launched in July 2019 by the ministries of research and agriculture with French laboratories handling prions. Submitted in October 2020, the report concluded on the regulatory compliance of the laboratories visited as well as the presence of a risk control culture within the research teams.
Research around prion proteins, with high public health issues, allows major advances in the understanding of the functioning of these infectious pathogens, and contributes to results that are transferable to other related degenerative diseases such as Alzheimer's and Alzheimer's diseases. Parkinson's.
At the level of each establishment, regular and transparent information will be provided to all the working communities concerned by this measure.
1 The disease Creutzfeldt-Jakob disease (CJD) is one of prion diseases - still called encephalopathies subacute spongiform transmitted(TSE) - of diseases rare, characterized by a degeneration rapid and fatal the system nervous central. They are caused by the accumulation in the brain of a normally expressed protein but poorly conformed - the prion protein - which leads to the formation of deleterious aggregates for neurons. For now , no treatment will allow to change the course of these diseases. It can be of origin sporadic , form the most frequent , original genetic or finally to form infectious following a contamination.
France issues moratorium on prion research after fatal brain disease strikes two lab workers
By Barbara CasassusJul. 28, 2021 , 4:35 AM
PARIS—Five public research institutions in France have imposed a 3-month moratorium on the study of prions—a class of misfolding, infectious proteins that cause fatal brain diseases—after a retired lab worker who handled prions in the past was diagnosed with Creutzfeldt-Jakob disease (CJD), the most common prion disease in humans. An investigation is underway to find out whether the patient, who worked at a lab run by the National Research Institute for Agriculture, Food and Environment (INRAE), contracted the disease on the job.
If so, it would be the second such case in France in the past few years. In June 2019, an INRAE lab worker named Émilie Jaumain died at age 33, 10 years after pricking her thumb during an experiment with prion-infected mice. Her family is now suing INRAE for manslaughter and endangering life; her illness had already led to tightened safety measures at French prion labs.
The aim of the moratorium, which affects nine labs, is to “study the possibility of a link with the [new patient’s] former professional activity and if necessary to adapt the preventative measures in force in research laboratories,” according to a joint press release issued by the five institutions yesterday.
“This is the right way to go in the circumstances,” says Ronald Melki, a structural biologist at a prion lab jointly operated by the French national research agency CNRS and the French Alternative Energies and Atomic Energy Commission (CEA). “It is always wise to ask questions about the whole working process when something goes wrong.” "The occurrence of these harsh diseases in two of our scientific colleagues clearly affects the whole prion community, which is a small 'familial' community of less than 1000 people worldwide," Emmanuel Comoy, deputy director of CEA's Unit of Prion Disorders and Related Infectious Agents, writes in an email to Science. Although prion research already has strict safety protocols, "it necessarily reinforces the awareness of the risk linked to these infectious agents," he says.
In Jaumain’s case, there is little doubt she was infected on the job, according to a paper published in The New England Journal of Medicine (NEJM) in 2020. She had variant CJD (vCJD), a form typically caused by eating beef contaminated with bovine spongiform encephalopathy (BSE), or mad cow disease. But Europe’s BSE outbreak ended after 2000 and vCJD virtually disappeared; the chance that someone of Jaumain’s age in France would contract food-borne vCJD is “negligible or non-existent,” according to the paper.
A scientist with inside knowledge says the new patient, a woman who worked at INRAE’s Host-Pathogen Interactions and Immunity group in Toulouse, is still alive. French authorities were apparently alerted to her diagnosis late last week. The press release suggests it’s not yet clear whether the new case is vCJD or “classic” CJD, which is not known to be caused by prions from animals. Classic CJD strikes an estimated one person per million. Some 80% of cases are sporadic, meaning they have no known cause, but others are genetic or contracted from infected human tissues during transplantations. The two types of CJD can only be distinguished through a postmortem examination of brain tissue.
Lab infections are known to occur with many pathogens, but exposure to CJD-causing prions is unusually risky because there are no vaccines or treatments and the condition is universally fatal. And whereas most infections reveal themselves within days or weeks, CJD’s average incubation period is about 10 years.
For Jaumain, who worked at INRAE’s Molecular Virology and Immunology Unit in Jouy-en-Josas, outside Paris, that long period of uncertainty began on 31 May 2010, when she stabbed her left thumb with a curved forceps while cleaning a cryostat—a machine that can cut tissues at very low temperatures—that she used to slice brain sections from transgenic mice infected with a sheep-adapted form of BSE. She pierced two layers of latex gloves and drew blood. “Émilie started worrying about the accident as soon as it had happened, and mentioned it to every doctor she saw,” says her widower, Armel Houel.
In November 2017, Jaumain developed a burning pain in her right shoulder and neck that worsened and spread to the right half of her body over the following 6 months, according to the NEJM paper. In January 2019, she became depressed and anxious, suffering memory impairment and hallucinations. “It was a descent into hell,” Houel says. She was diagnosed with “probable vCJD” in mid-March of that year and died 3 months later. A postmortem confirmed the diagnosis.
“The occurrence of these harsh diseases in two of our scientific colleagues clearly affects the whole prion community.” Emmanuel Comoy, French Alternative Energies and Atomic Energy Commission
INRAE only recently admitted the likely link between Jaumain’s illness and the accident. “We recognize, without ambiguity, the hypothesis of a correlation between Emilie Jaumain-Houel’s accident … and her infection with vCJD,” INRAE chair and CEO Philippe Mauguin wrote in a 24 June letter to an association created by friends and colleagues to publicize Jaumain’s case and lobby for improvements in lab safety. (Science has obtained a copy of the letter, which has not been made public.)
Jaumain’s family has filed both criminal charges and an administrative suit against INRAE, alleging a range of problems at Jaumain’s lab. She had not been trained in handling dangerous prions or responding to accidents and did not wear both metal mesh and surgical gloves, as she was supposed to, says Julien Bensimhon, the family’s lawyer. The thumb should have been soaked in a bleach solution immediately, which did not happen, Bensimhon adds.
Independent reports by a company specializing in occupational safety and by government inspectors have found no safety violations at the lab; one of them said there was a “strong culture” of risk management. (Bensimhon calls the reports “biased.”)
The government inspectors’ report concluded that Jaumain’s accident was not unique, however. There had been at least 17 accidents among the 100 or so scientists and technicians in France working with prions in the previous decade, five of whom stabbed or cut themselves with contaminated syringes or blades. Another technician at the same lab had a fingerprick accident with prions in 2005, but has not developed vCJD symptoms so far, Bensimhon says. “It is shocking that no precautionary measures were taken then to ensure such an accident never happened again,” he says.
In Italy, too, the last person to die of vCJD, in 2016, was a lab worker with exposure to prion-infected brain tissue, according to last year’s NEJM paper, although an investigation did not find evidence of a lab accident. That patient and the lab they worked at have not been identified.
After Jaumain’s diagnosis, “We contacted all the research prion labs in France to suggest they check their safety procedures and remind staff about the importance of respecting them,” says Stéphane Haïk, a neuroscientist at the Paris Brain Institute at Pitié-Salpêtrière Hospital who helped diagnose Jaumain and is the corresponding author on the paper. Many labs tightened procedures, according to the government inspectors' report, for instance by introducing plastic scissors and scalpels, which are disposable and less sharp, and bite and cut-resistant gloves. A team of experts from the five research agencies is due to submit proposals for a guide to good practice in prion research to the French government at the end of this year.
The scientific community has long recognized that handling prions is dangerous and an occupational risk for neuropathologists, says neuropathologist Adriano Aguzzi of the University of Zurich. Aguzzi declined to comment on the French CJD cases, but told Science his lab never handles human or bovine prions for research purposes, only for diagnostics. “We conduct research only on mouse-adapted sheep prions, which have never been shown to be infectious to humans,” Aguzzi says. In a 2011 paper, his team reported that prions can spread through aerosols, at least in mice, which “may warrant re-thinking on prion biosafety guidelines in research and diagnostic laboratories,” they wrote. Aguzzi says he was “totally shocked” by the finding and introduced safety measures to prevent aerosol spread at his own lab, but the paper drew little attention elsewhere.
The moratorium will "obviously" cause delays in research, but given the very long incubation periods in prion diseases, the impact of a 3-month hiatus will be limited, Comoy says. His research team at CEA also works on other neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, and will shift some of its efforts to those.
Although Jaumain’s diagnosis upset many in the field, it hasn't led to an exodus among researchers in France, Haïk says: “I know of only one person who resigned because they were so worried.”
With reporting by Martin Enserink.
Posted in: EuropeHealthScientific Community
doi:10.1126/science.abl6587
Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure
Variant Creutzfeldt–Jakob disease was identified in a technician who had cut her thumb while handling brain sections of mice infected with adapted BSE 7.5 years earlier. The long incubation period was similar to that of the transfusion-transmitted form of the disease.
Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure
TO THE EDITOR:
We report a case of variant Creutzfeldt–Jakob disease (CJD) that was plausibly related to accidental occupational exposure in a technician who had handled murine samples contaminated with the agent that causes bovine spongiform encephalopathy (BSE) 7.5 years earlier.
In May 2010, when the patient was 24 years of age, she worked in a prion research laboratory, where she handled frozen sections of brain of transgenic mice that overexpressed the human prion protein with methionine at codon 129. The mice had been infected with a sheep-adapted form of BSE. During this process, she stabbed her thumb through a double pair of latex gloves with the sharp ends of a curved forceps used to handle the samples. Bleeding was noted at the puncture site.
In November 2017, she began having burning pain in the right shoulder and neck. The pain worsened and spread to the right half of her body during the following 6 months. In November 2018, an examination of a sample of cerebrospinal fluid (CSF) obtained from the patient was normal. Magnetic resonance imaging (MRI) of the brain showed a slight increase in the fluid-attenuated inversion recovery (FLAIR) signal in the caudates and thalami (Fig. S1A and S1B in the Supplementary Appendix, available with the full text of this letter at NEJM.org). In January 2019, she became depressed and anxious and had memory impairment and visual hallucinations. There was hypertonia on the right side of her body. At that time, an analysis of CSF for 14-3-3 protein was negative. In March 2019, MRI showed an increased FLAIR signal in pulvinar and dorsomedial nuclei of thalami (Fig. S1C through S1E).
Figure 1.
Detection of Abnormal Prion Protein in Biologic Fluid Samples and Postmortem Findings.
The patient was found to be homozygous for methionine at codon 129 of the prion protein gene without mutation. An analysis of a sample of CSF on real-time quaking-induced conversion analysis was negative for a diagnosis of sporadic CJD. However, an analysis of plasma and CSF by means of protein misfolding cyclic amplification was positive for the diagnosis of variant CJD (Figure 1A and 1B). The patient died 19 months after the onset of symptoms. Neuropathological examination confirmed the diagnosis of variant CJD (Figure 1C and 1D). Western blot analysis showed the presence of type 2B protease-resistant prion protein in all sampled brain areas. The clinical characteristics of the patient and the postmortem neuropathological features were similar to those observed in 27 patients with variant CJD who had previously been reported in France.1 (Additional details are provided in the Supplementary Appendix.)
There are two potential explanations for this patient’s condition. Oral transmission from contaminated cattle products cannot be ruled out because the patient was born at the beginning of the French BSE outbreak in cattle. However, the last two patients who had confirmed variant CJD with methionine homozygosity at codon 129 in France and the United Kingdom died in 2014 and 2013, respectively, which makes oral transmission unlikely. In France, the risk of variant CJD in 2019 was negligible or nonexistent in the post-1969 birth cohort.2
Percutaneous exposure to prion-contaminated material is plausible in this patient, since the prion strain that she had handled was consistent with the development of variant CJD.3 The 7.5-year delay between the laboratory accident and her clinical symptoms is congruent with the incubation period in the transfusion-transmitted form of the disease. The ability of this strain to propagate through the peripheral route has been documented, and experimental studies with scrapie strains have shown that scarification and subcutaneous inoculation are effective routes.4,5 The last known Italian patient with variant CJD, who died in 2016, had had occupational contact with BSE-infected brain tissues, although subsequent investigation did not disclose a laboratory accident (Pocchiari M, Italian Registry of CJD: personal communication). Thus, the last two cases of variant CJD outside the United Kingdom have been associated with potential occupational exposure. Such cases highlight the need for improvements in the prevention of transmission of variant CJD and other prions that can affect humans in the laboratory and neurosurgery settings, as outlined in the Supplementary Appendix.
Jean-Philippe Brandel, M.D. Assistance Publique–Hôpitaux de Paris, Paris, France
M. Bustuchina Vlaicu, M.D. Groupe Hospitalier Nord-Essonne, Orsay, France
Audrey Culeux, B.Sc. INSERM Unité 1127, Paris, France
Maxime Belondrade, M.Sc. Daisy Bougard, Ph.D. Etablissement Français du Sang, Montpellier, France
Katarina Grznarova, Ph.D. Angeline Denouel, M.Sc. INSERM Unité 1127, Paris, France
Isabelle Plu, M.D. Elodie Bouaziz-Amar, Pharm.D., Ph.D. Danielle Seilhean, M.D., Ph.D. Assistance Publique–Hôpitaux de Paris, Paris, France
Michèle Levasseur, M.D. Groupe Hospitalier Nord-Essonne, Orsay, France
Stéphane Haïk, M.D., Ph.D. INSERM Unité 1127, Paris, France stephane.haik@upmc.fr
Supported by a grant (ANR-10-IAIHU-06) from Programme d’Investissements d’Avenir and Santé Publique France.
Disclosure forms provided by the authors are available with the full text of this letter at NEJM.org.
5 References
July 2, 2020
N Engl J Med 2020; 383:83-85
DOI: 10.1056/NEJMc2000687
Metrics
34 year old Doctor Orthopedic Surgeon dies from CJD
Dr. Adam Thomas Dialectos
1987 - 2021
BORN
April 29, 1987
DIED
June 21, 2021
FUNERAL HOME
Bean Funeral Homes & Crematory Inc
1605 Rockland St
Reading, PA 19604
UPCOMING SERVICE
Visitation
Jun, 24 2021
9:00a.m. - 11:00a.m.
Saints Constantine & Helen Greek Orthodox Church
Send Flowers
Share
On Monday June 21, 2021, Dr. Adam Thomas Dialectos, loving husband, father, son, brother, uncle, Nouno, friend at the age of 34. Adam was born on April 29, 1987 in Reading, PA to Athan and Gretchen Dialectos. Adam was a 2005 graduate of Governor Mifflin High School, before receiving his degree in Health Sciences from James Madison University in 2009. Adam attended Philadelphia College of Osteopathic Medicine for medical school and his subsequent residency in orthopedic surgery. Adam was completing his Spine Surgery Fellowship at New England Baptist Hospital in Boston, Massachusetts. On February 7, 2019 Adam married the love of his life and girlfriend of 12 years, Lindsey (Schuler) Dialectos. They brought a beautiful baby boy into this world on January 6, 2021, Athananosis Adam Dialectos. Adam’s passion in life was unceasingly seeking to help others, emphasized by his desire to be a surgeon— a decision he made in his early elementary years. Adam continued this love of medicine throughout his life, which led to his achieving of the Henrietta and Jack Avart Memorial Award in 2019, awarded to the Orthopedic surgery resident who exhibited unparalleled excellence in their field during the residency program. This passion to learn, teach and support was truly understood through the patients whose lives Adam touched. When it came to his patients and coworkers, there was never a job too small for Adam. Those who knew Adam saw his personality shine through in so many other aspects of his life. Adam loved traveling. Some of his most memorable trips were with his wife, and countless snowboard trips with his brother, family, and friends. Adam loved everyone he was around; he loved and was loved by so many. Adam was truly one in a million. Adam is survived by his loving wife, Lindsey, and their son, Athan Adam; His father and mother, Athan and Gretchen; His brother Jordan and sister-in-law Megan, and their daughter Livia, Adam’s Goddaughter. His sister, Rachel, and her significant other, Bo Wagner. Furthermore, Adam is survived by his Yiayia, Joanne Dialectos, wife of the late George Dialectos; his Pop Pop, Donald Harford, husband of the late Nancy Servent; his Aunt Angel and Uncle Scott Helm; his Aunt Kelly and Uncle Darrell Markley. Adam was preceded in death by his Aunt Maria and Uncle Bob Care. Funeral Service will be held at Saints Constantine & Helen Greek Orthodox Church, 1001 East Wyomissing Blvd. Reading on Thursday June 24th. Father Theodore Petrides and Father Thomas L. Pappalas will officiate. Interment will follow at Charles Evans Cemetery. The family will receive relatives and friends at Saints Constantine & Helen Greek Orthodox Church from 9:00am to 11:00am with services beginning at 11:00. In lieu of flowers, contributions may be made to the CJD Foundation at 3634 West Market Street Suite 110 Akron, Ohio 44333 or cjdfoundation.org in remembrance of Dr. Adam Dialectos. Donations may also be made to Saints Constantine & Helen Greek Orthodox Church. Bean Funeral Home, 1605 Rockland Street, Hampden Heights, is in charge of arrangements and online condolences may be made at www.beanfuneralhomes.com.
To plant trees in memory, please visit our Sympathy Store.
Published by Reading Eagle from Jun. 22 to Jun. 24, 2021.
Our sincere condolences to the Family and Friends of Dr. Adam Thomas Dialectos.
I can't help but ponder, as a Orthopedic Surgeon, Spine Surgery Fellowship, and what the good Doctors work curtailed, i can't help but think this is a potential case of iatrogenic CJD. surgery on humans, i would imagine cadavers as well.
all iatrogenic cjd is, is sporadic cjd, before the iatrogenic event is discovered, traced back, provern, documented, put into the academic domain, and then finally the public domain, this very seldom happens, thus problem solved, it's all sporadic cjd. ...terry
least we forget...
*** Transmission of Creutzfeldt-Jakob disease to a chimpanzee by electrodes contaminated during neurosurgery ***
Gibbs CJ Jr, Asher DM, Kobrine A, Amyx HL, Sulima MP, Gajdusek DC.
Laboratory of Central Nervous System Studies, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892. Stereotactic multicontact electrodes used to probe the cerebral cortex of a middle aged woman with progressive dementia were previously implicated in the accidental transmission of Creutzfeldt-Jakob disease (CJD) to two younger patients. The diagnoses of CJD have been confirmed for all three cases. More than two years after their last use in humans, after three cleanings and repeated sterilisation in ethanol and formaldehyde vapour, the electrodes were implanted in the cortex of a chimpanzee. Eighteen months later the animal became ill with CJD. This finding serves to re-emphasise the potential danger posed by reuse of instruments contaminated with the agents of spongiform encephalopathies, even after scrupulous attempts to clean them.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8006664&dopt=Abstract
Saturday, December 18, 2021
Direct neural transmission of vCJD/BSE in macaque after finger incision
Tuesday, November 30, 2021
Second death in France in a laboratory working on prions
Second lab worker with deadly prion disease prompts research pause in France
A lab worker died of prion disease in 2019, nine years after a lab accident.
BETH MOLE - 7/29/2021, 5:16 PM
A 2020 paper published in the New England Journal of Medicine left little doubt that Jaumain had been infected on the job. She had variant CJD, but since Europe’s ‘mad cow’ outbreak ended after 2000 and the disease virtually disappeared, the paper said it was virtually impossible for someone her age in France to contract food-borne vCJD.
Science also said two independent reports – one by government inspectors – had found no safety violations at the lab where Jaumain worked. The press release also noted that the inspectors concluded there was “the presence of a risk control culture within the research teams”. The Jaumain family’s lawyer called the neutrality of the reports into question, however.
At the same time, the government inspectors’ report also revealed that there had been at least 17 accidents among the 100 or so scientists and technicians in France working with prions in the previous decade, raising concerns about how effective this risk control culture is. Five of these occurred when workers “stabbed or cut themselves with contaminated syringes or blades”.
Wednesday, July 28, 2021
France issues moratorium on prion research after fatal brain disease strikes two lab workers
Wednesday, July 28, 2021
France issues moratorium on prion research after fatal brain disease strikes two lab workers
SATURDAY, AUGUST 01, 2020
Sporadic Creutzfeldt-Jakob Disease among Physicians, Germany, 1993–2018 high proportion of physicians with sCJD were surgeons
SUNDAY, JULY 19, 2020
Joseph J. Zubak Orthopaedic surgeon passed away Monday, July 6, 2020, Creutzfeldt-Jakob Disease (CJD)
Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure
Variant Creutzfeldt–Jakob disease was identified in a technician who had cut her thumb while handling brain sections of mice infected with adapted BSE 7.5 years earlier. The long incubation period was similar to that of the transfusion-transmitted form of the disease.
THURSDAY, JULY 02, 2020
Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure
Thursday, July 29, 2021
TSE PRION OCCUPATIONAL EXPOSURE VIA ANIMAL OR HUMAN, iatrogenic transmission, nvCJD or sCJD, what if?
Deep Throat to Singeltary BSE Mad Cow 2001 to 2023
I remember what “deep throat” told me about Scrapie back around 2001, I never forgot, and it seems it’s come to pass;
***> Confidential!!!!
***> As early as 1992-3 there had been long studies conducted on small pastures containing scrapie infected sheep at the sheep research station associated with the Neuropathogenesis Unit in Edinburgh, Scotland. Whether these are documented...I don't know. But personal recounts both heard and recorded in a daily journal indicate that leaving the pastures free and replacing the topsoil completely at least 2 feet of thickness each year for SEVEN years....and then when very clean (proven scrapie free) sheep were placed on these small pastures.... the new sheep also broke out with scrapie and passed it to offspring. I am not sure that TSE contaminated ground could ever be free of the agent!! A very frightening revelation!!!
---end personal email---end...tss
(I never knew who this person was, but got me into the U.S. BSE Emergency 50 State conference call back 2001, and we corresponded for years about BSE TSE Prion, have not heard from in over a decade, but they were on the inside looking out. You can believe this or not, but this was real, i don’t make this stuff up…plus my endeavors to get those 1 million cattle tested for BSE failed. There was an ENHANCED BSE SURVEILLANCE put forth after 2003, we pushed for it, but it was abruptly shut down after the atypical BSE cases were popping up…a bit of history for anyone interested…terry)
DEEP THROAT TO TSS 2000-2001 (take these old snips of emails with how ever many grains of salt you wish. ...tss)
The most frightening thing I have read all day is the report of Gambetti's finding of a new strain of sporadic cjd in young people...Dear God, what in the name of all that is holy is that!!! If the US has different strains of scrapie.....why???? than the UK...then would the same mechanisms that make different strains of scrapie here make different strains of BSE...if the patterns are different in sheep and mice for scrapie.....could not the BSE be different in the cattle, in the mink, in the humans.......I really think the slides or tissues and everything from these young people with the new strain of sporadic cjd should be put up to be analyzed by many, many experts in cjd........bse.....scrapie Scrape the damn slide and put it into mice.....wait.....chop up the mouse brain and and spinal cord........put into some more mice.....dammit amplify the thing and start the damned research.....This is NOT rocket science...we need to use what we know and get off our butts and move....the whining about how long everything takes.....well it takes a whole lot longer if you whine for a year and then start the research!!!
Not sure where I read this but it was a recent press release or something like that: I thought I would fall out of my chair when I read about how there was no worry about infectivity from a histopath slide or tissues because they are preserved in formic acid, or formalin or formaldehyde.....for God's sake........ Ask any pathologist in the UK what the brain tissues in the formalin looks like after a year.......it is a big fat sponge...the agent continues to eat the brain ......you can't make slides anymore because the agent has never stopped........and the old slides that are stained with Hemolysin and Eosin......they get holier and holier and degenerate and continue...what you looked at 6 months ago is not there........Gambetti better be photographing every damned thing he is looking at.....
Okay, you need to know. You don't need to pass it on as nothing will come of it and there is not a damned thing anyone can do about it. Don't even hint at it as it will be denied and laughed at.......... USDA is gonna do as little as possible until there is actually a human case in the USA of the nvcjd........if you want to move this thing along and shake the earth....then we gotta get the victims families to make sure whoever is doing the autopsy is credible, trustworthy, and a saint with the courage of Joan of Arc........I am not kidding!!!! so, unless we get a human death from EXACTLY the same form with EXACTLY the same histopath lesions as seen in the UK nvcjd........forget any action........it is ALL gonna be sporadic!!! And, if there is a case.......there is gonna be every effort to link it to international travel, international food, etc. etc. etc. etc. etc. They will go so far as to find out if a sex partner had ever traveled to the UK/europe, etc. etc. .... It is gonna be a long, lonely, dangerous twisted journey to the truth. They have all the cards, all the money, and are willing to threaten and carry out those threats....and this may be their biggest downfall...
Thanks as always for your help. (Recently had a very startling revelation from a rather senior person in government here..........knocked me out of my chair........you must keep pushing. If I was a power person....I would be demanding that there be a least a million bovine tested as soon as possible and agressively seeking this disease. The big players are coming out of the woodwork as there is money to be made!!! In short: "FIRE AT WILL"!!! for the very dumb....who's "will"! "Will be the burden to bare if there is any coverup!"
again it was said years ago and it should be taken seriously....BSE will NEVER be found in the US! As for the BSE conference call...I think you did a great service to freedom of information and making some people feign integrity...I find it scary to see that most of the "experts" are employed by the federal government or are supported on the "teat" of federal funds. A scary picture! I hope there is a confidential panel organized by the new government to really investigate this thing.
You need to watch your back........but keep picking at them.......like a buzzard to the bone...you just may get to the truth!!! (You probably have more support than you know. Too many people are afraid to show you or let anyone else know. I have heard a few things myself... you ask the questions that everyone else is too afraid to ask.)
U.S. 50 State Emergency BSE Conference Call 2001
Singeltary Comment Docket No: 2002N-0273 (formerly Docket No. 02N-0273)
MY comments/questions are as follows ;
1. SINCE the first Harvard BSE Risk Assessment was so flawed and fraught with error after the PEER REVIEW assessment assessed this fact, how do you plan on stopping this from happening again, will there be another peer review with top TSE Scientist, an impartial jury so-to-speak, to assess this new and updated Harvard BSE/TSE risk assessment and will this assessment include the Atypical TSE and SRM issues ?
*** Suppressed peer review of Harvard study October 31, 2002 ***
2. WITH A RECENT NATION WIDE MAD COW FEED BAN RECALL in the past few months that consisted of some 10,878.06 TONS, then another Mad Cow feed ban warning letter in May, IT should seem prudent to ask why our feed bans continue to fail in 2006, and continue to fail today ?
snip...see full text;
Singeltary Full Comments Submissions;
FSIS, HARVARD, REPLY TO SINGELTARY
Document APHIS-2023-0027-0001 BSE Singeltary Comment Submission
see full submission;
Specified Risk Materials DOCKET NUMBER Docket No. FSIS-2022-0027 Singeltary Submission Attachment
APHIS Concurrence With OIE Risk Designation for Bovine Spongiform Encephalopathy [Docket No. APHIS-2018-0087] Singeltary Submission Comment from Terry Singeltary Posted by the Animal and Plant Health Inspection Service on Jun 19, 2019
Control of Chronic Wasting Disease OMB Control Number: 0579-0189APHIS-2021-0004 Singeltary Submission
Docket No. APHIS-2018-0011 Chronic Wasting Disease Herd Certification
APHIS Indemnity Regulations [Docket No. APHIS-2021-0010] RIN 0579-AE65 Singeltary Comment Submission
Comment from Singeltary Sr., Terry
Posted by the Animal and Plant Health Inspection Service on Sep 8, 2022
Docket No. FDA-2003-D-0432 (formerly 03D-0186) Use of Material from Deer and Elk in Animal Feed
PUBLIC SUBMISSION
Comment from Terry Singeltary Sr.
Posted by the Food and Drug Administration on May 17, 2016 Comment
Docket No. FDA-2003-D-0432 (formerly 03D-0186) Use of Material from Deer and Elk in Animal Feed Singeltary Submission
FRIDAY, APRIL 07, 2023
Case report: Two clusters of Creutzfeldt-Jakob disease cases within 1 year in West Michigan
MONDAY, APRIL 24, 2023
2023 CDC REPORTS CJD TSE Prion 5 cases per million in persons 55 years of age or older
THE PATHOLOGICAL PROTEIN
Hardcover, 304 pages plus photos and illustrations. ISBN 0-387-95508-9
June 2003
BY Philip Yam
CHAPTER 14 LAYING ODDS
Answering critics like Terry Singeltary, who feels that the U.S. under- counts CJD, Schonberger conceded that the current surveillance system has errors but stated that most of the errors will be confined to the older population.
reviewed medical records for CJD cases between 1991 and 1995. Comparing the actively garnered data with the death certificate infor-mation showed that “we miss about 14 percent,” said CDC epidemiolo-gist Lawrence Schonberger.
RE-Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United States
Terry S. Singeltary, retired (medically), CJD WATCH
Submitted March 26, 2003
I lost my mother to hvCJD (Heidenhain Variant CJD). I would like to comment on the CDC's attempts to monitor the occurrence of emerging forms of CJD. Asante, Collinge et al [1] have reported that BSE transmission to the 129-methionine genotype can lead to an alternate phenotype that is indistinguishable from type 2 PrPSc, the commonest sporadic CJD. However, CJD and all human TSEs are not reportable nationally. CJD and all human TSEs must be made reportable in every state and internationally. I hope that the CDC does not continue to expect us to still believe that the 85%+ of all CJD cases which are sporadic are all spontaneous, without route/source. We have many TSEs in the USA in both animal and man. CWD in deer/elk is spreading rapidly and CWD does transmit to mink, ferret, cattle, and squirrel monkey by intracerebral inoculation. With the known incubation periods in other TSEs, oral transmission studies of CWD may take much longer. Every victim/family of CJD/TSEs should be asked about route and source of this agent. To prolong this will only spread the agent and needlessly expose others. In light of the findings of Asante and Collinge et al, there should be drastic measures to safeguard the medical and surgical arena from sporadic CJDs and all human TSEs. I only ponder how many sporadic CJDs in the USA are type 2 PrPSc?
Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United States 2003 revisited 2009
August 10, 2009
Greetings,
I would like to submit a review of past CJD surveillance in the USA, and the urgent need to make all human TSE in the USA a reportable disease, in every state, of every age group, and to make this mandatory immediately without further delay. The ramifications of not doing so will only allow this agent to spread further in the medical, dental, surgical arena's. North America seems to have the most species with documented Transmissible Spongiform Encephalopathy's, most all of which have been rendered and fed back to food producing animals and to humans for years. If you look at the statistics, sporadic CJD seems to be rising in the USA, and has been, with atypical cases of the sCJD. I find deeply disturbing in the year of 2009, that Human Transmissible Spongiform Encephalopathy of any strain and or phenotype, of all age groups, and I stress all age groups, because human TSE's do not know age, and they do not know borders. someone 56 years old, that has a human TSE, that has surgery, can pass this TSE agent on i.e. friendly fire, and or passing it forward, and there have been documented nvCJD in a 74 year old. Remembering also that only sporadic CJD has been documented to transmit via iatrogenic routes, until recently with the 4 cases of blood related transmission, of which the origin is thought to be nvCJD donors. However most Iatrogenic CJD cases are nothing more than sporadic CJD, until the source is proven, then it becomes Iatrogenic. An oxymoron of sorts, because all sporadic CJD is, are multiple forms, or strains, or phenotypes of Creutzfeldt Jakob Disease, that the route and source and species have not been confirmed and or documented. When will the myth of the UKBSEnvCJD only theory be put to bed for good. This theory in my opinion, and the following there from, as the GOLD STANDARD, has done nothing more than help spread this agent around the globe. Politics and money have caused the terrible consequences to date, and the fact that TSEs are a slow incubating death, but a death that is 100% certain for those that are exposed and live long enough to go clinical. once clinical, there is no recourse, to date.
But, while sub-clinical, how many can one exposed human infect?
Can humans exposed to CWD and scrapie strains pass it forward as some form of sporadic CJD in the surgical and medical arenas?
why must we wait decades and decades to prove this point, only to expose millions needlessly, only for the sake of the industries involved?
would it not have been prudent from the beginning to just include all TSE's, and rule them out from there with transmission studies and change policies there from, as opposed to doing just the opposite?
The science of TSE's have been nothing more than a political circus since the beginning, and for anyone to still believe in this one strain, one group of bovines, in one geographical location, with only one age group of human TSE i.e. nvCJD myth, for anyone to believe this today only enhances to spreading of these human and animal TSE's. This is exactly why we have been in this quagmire.
The ones that believe that there is a spontaneous CJD in 85%+ of all cases of human TSE, and the ones that do not believe that cattle can have this same phenomenon, are two of the same, the industry, and so goes the political science aspect of this tobacco and or asbestos scenario i.e. follow the money. I could go into all angles of this man made nightmare, the real facts and science, for instance, the continuing rendering technology and slow cooking with low temps that brewed this stew up, and the fact that THE USA HAD THIS TECHNOLOGY FIRST AND SHIPPED IT TO THE U.K. SOME 5 YEARS BEFORE THE U.S. STARTED USING THE SAME TECHNOLOGY, to save on fuel cost. This is what supposedly amplified the TSE agent via sheep scrapie, and spread via feed in the U.K. bovine, and other countries exporting the tainted product. BUT most everyone ignores this fact, and the fact that the U.S. has been recycling more TSE, from more species with TSEs, than any other country documented, but yet, it's all spontaneous, and the rise in sporadic CJD in the U.S. is a happenstance of bad luck ??? I respectfully disagree. To top that all off, the infamous BSE-FIREWALL that the USDA always brags about was nothing more than ink on paper, and I can prove this. YOU can ignore it, but this is FACT (see source, as late as 2007, in one recall alone, some 10,000,000 MILLION POUNDS OF BANNED MAD COW FEED WENT OUT INTO COMMERCE TO BE FED OUT, and most was never recovered. This was banned blood laced, meat and bone meal. 2006 was a banner year for banned mad cow protein going into commerce in the U.S. (see source of FDA feed ban warning letter below). I stress that the August 4, 1997 USA mad cow feed ban and this infamous BSE firewall, was nothing more than ink on paper, it was never enforceable.
I propose that the current diagnostic criteria for human TSEs only enhances and helps the spreading of human TSE from the continued belief of the UKBSEnvCJD only theory in 2009. With all the science to date refuting it, to continue to validate this old myth, will only spread this TSE agent through a multitude of potential routes and sources i.e. consumption, medical i.e., surgical, blood, dental, endoscopy, optical, nutritional supplements, cosmetics etc. I propose as with Aguzzi, Asante, Collinge, Caughey, Deslys, Dormont, Gibbs, Gajdusek, Ironside, Manuelidis, Marsh, et al and many more, that the world of TSE Transmissible Spongiform Encephalopathy is far from an exact science, but there is enough proven science to date that this myth should be put to rest once and for all, and that we move forward with a new classification for human and animal TSE that would properly identify the infected species, the source species, and then the route. This would further have to be broken down to strain of species and then the route of transmission would further have to be broken down. Accumulation and Transmission are key to the threshold from sub- clinical to clinical disease, and key to all this, is to stop the amplification and transmission of this agent, the spreading of, no matter what strain. In my opinion, to continue with this myth that the U.K. strain of BSE one strain TSE in cows, and the nv/v CJD one strain TSE humans, and the one geographical location source i.e. U.K., and that all the rest of human TSE are just one single strain i.e. sporadic CJD, a happenstance of bad luck that just happens due to a twisted protein that just twisted the wrong way, IN 85%+ OF ALL HUMAN TSEs, when to date there are 6 different phenotypes of sCJD, and growing per Gambetti et al, and that no other animal TSE transmits to humans ??? With all due respect to all Scientist that believe this, I beg to differ. To continue with this masquerade will only continue to spread, expose, and kill, who knows how many more in the years and decades to come. ONE was enough for me, My Mom, hvCJD i.e. Heidenhain Variant CJD, DOD 12/14/97 confirmed, which is nothing more than another mans name added to CJD, like CJD itself, Jakob and Creutzfeldt, or Gerstmann-Straussler-Scheinker syndrome, just another CJD or human TSE, named after another human. WE are only kidding ourselves with the current diagnostic criteria for human and animal TSE, especially differentiating between the nvCJD vs the sporadic CJD strains and then the GSS strains and also the FFI fatal familial insomnia strains or the ones that mimics one or the other of those TSE? Tissue infectivity and strain typing of the many variants of the human and animal TSEs are paramount in all variants of all TSE. There must be a proper classification that will differentiate between all these human TSE in order to do this. With the CDI and other more sensitive testing coming about, I only hope that my proposal will some day be taken seriously. ...
please see history, and the ever evolving TSE science to date ;
Saturday, June 13, 2009
Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United States 2003 revisited 2009
Singeltary 2000
BMJ 2000; 320 doi: https://doi.org/10.1136/bmj.320.7226.8/b (Published 01 January 2000) Cite this as: BMJ 2000;320:8
02 January 2000 Terry S Singeltary retired
Rapid Response:
U.S. Scientist should be concerned with a CJD epidemic in the U.S., as well...
In reading your short article about 'Scientist warn of CJD epidemic' news in brief Jan. 1, 2000. I find the findings in the PNAS old news, made famous again. Why is the U.S. still sitting on their butts, ignoring the facts? We have the beginning of a CJD epidemic in the U.S., and the U.S. Gov. is doing everything in it's power to conceal it.
The exact same recipe for B.S.E. existed in the U.S. for years and years. In reading over the Qualitative Analysis of BSE Risk Factors-1, this is a 25 page report by the USDA:APHIS:VS. It could have been done in one page. The first page, fourth paragraph says it all;
"Similarities exist in the two countries usage of continuous rendering technology and the lack of usage of solvents, however, large differences still remain with other risk factors which greatly reduce the potential risk at the national level."
Then, the next 24 pages tries to down-play the high risks of B.S.E. in the U.S., with nothing more than the cattle to sheep ratio count, and the geographical locations of herds and flocks. That's all the evidence they can come up with, in the next 24 pages.
Something else I find odd, page 16;
"In the United Kingdom there is much concern for a specific continuous rendering technology which uses lower temperatures and accounts for 25 percent of total output. This technology was _originally_ designed and imported from the United States. However, the specific application in the production process is _believed_ to be different in the two countries."
A few more factors to consider, page 15;
"Figure 26 compares animal protein production for the two countries. The calculations are based on slaughter numbers, fallen stock estimates, and product yield coefficients. This approach is used due to variation of up to 80 percent from different reported sources. At 3.6 million tons, the United States produces 8 times more animal rendered product than the United Kingdom."
"The risk of introducing the BSE agent through sheep meat and bone meal is more acute in both relative and absolute terms in the United Kingdom (Figures 27 and 28). Note that sheep meat and bone meal accounts for 14 percent, or 61 thousand tons, in the United Kingdom versus 0.6 percent or 22 thousand tons in the United States. For sheep greater than 1 year, this is less than one-tenth of one percent of the United States supply."
"The potential risk of amplification of the BSE agent through cattle meat and bone meal is much greater in the United States where it accounts for 59 percent of total product or almost 5 times more than the total amount of rendered product in the United Kingdom."
Considering, it would only take _one_ scrapie infected sheep to contaminate the feed. Considering Scrapie has run rampant in the U.S. for years, as of Aug. 1999, 950 scrapie infected flocks. Also, Considering only one quarter spoonful of scrapie infected material is lethal to a cow.
Considering all this, the sheep to cow ration is meaningless. As I said, it's 24 pages of B.S.e.
To be continued...
Terry S. Singeltary Sr. Bacliff, Texas USA
Competing interests: No competing interests
doi:10.1016/S1473-3099(03)00715-1 Copyright © 2003 Published by Elsevier Ltd. Newsdesk
Tracking spongiform encephalopathies in North America
Xavier Bosch
Available online 29 July 2003.
Volume 3, Issue 8, August 2003, Page 463
Volume 3, Number 8 01 August 2003
Newsdesk
Tracking spongiform encephalopathies in North America
Xavier Bosch
My name is Terry S Singeltary Sr, and I live in Bacliff, Texas. I lost my mom to hvCJD (Heidenhain variant CJD) and have been searching for answers ever since. What I have found is that we have not been told the truth. CWD in deer and elk is a small portion of a much bigger problem.
49-year-old Singeltary is one of a number of people who have remained largely unsatisfied after being told that a close relative died from a rapidly progressive dementia compatible with spontaneous Creutzfeldt-Jakob disease (CJD). So he decided to gather hundreds of documents on transmissible spongiform encephalopathies (TSE) and realised that if Britons could get variant CJD from bovine spongiform encephalopathy (BSE), Americans might get a similar disorder from chronic wasting disease (CWD) the relative of mad cow disease seen among deer and elk in the USA. Although his feverish search did not lead him to the smoking gun linking CWD to a similar disease in North American people, it did uncover a largely disappointing situation.
Singeltary was greatly demoralised at the few attempts to monitor the occurrence of CJD and CWD in the USA. Only a few states have made CJD reportable. Human and animal TSEs should be reportable nationwide and internationally, he complained in a letter to the Journal of the American Medical Association (JAMA 2003; 285: 733). I hope that the CDC does not continue to expect us to still believe that the 85% plus of all CJD cases which are sporadic are all spontaneous, without route or source.
Until recently, CWD was thought to be confined to the wild in a small region in Colorado. But since early 2002, it has been reported in other areas, including Wisconsin, South Dakota, and the Canadian province of Saskatchewan. Indeed, the occurrence of CWD in states that were not endemic previously increased concern about a widespread outbreak and possible transmission to people and cattle.
To date, experimental studies have proven that the CWD agent can be transmitted to cattle by intracerebral inoculation and that it can cross the mucous membranes of the digestive tract to initiate infection in lymphoid tissue before invasion of the central nervous system. Yet the plausibility of CWD spreading to people has remained elusive.
Part of the problem seems to stem from the US surveillance system. CJD is only reported in those areas known to be endemic foci of CWD. Moreover, US authorities have been criticised for not having performed enough prionic tests in farm deer and elk.
Although in November last year the US Food and Drug Administration issued a directive to state public-health and agriculture officials prohibiting material from CWD-positive animals from being used as an ingredient in feed for any animal species, epidemiological control and research in the USA has been quite different from the situation in the UK and Europe regarding BSE.
Getting data on TSEs in the USA from the government is like pulling teeth, Singeltary argues. You get it when they want you to have it, and only what they want you to have.
Norman Foster, director of the Cognitive Disorders Clinic at the University of Michigan (Ann Arbor, MI, USA), says that current surveillance of prion disease in people in the USA is inadequate to detect whether CWD is occurring in human beings; adding that, the cases that we know about are reassuring, because they do not suggest the appearance of a new variant of CJD in the USA or atypical features in patients that might be exposed to CWD. However, until we establish a system that identifies and analyses a high proportion of suspected prion disease cases we will not know for sure. The USA should develop a system modelled on that established in the UK, he points out.
Ali Samii, a neurologist at Seattle VA Medical Center who recently reported the cases of three hunters two of whom were friends who died from pathologically confirmed CJD, says that at present there are insufficient data to claim transmission of CWD into humans; adding that [only] by asking [the questions of venison consumption and deer/elk hunting] in every case can we collect suspect cases and look into the plausibility of transmission further. Samii argues that by making both doctors and hunters more aware of the possibility of prions spreading through eating venison, doctors treating hunters with dementia can consider a possible prion disease, and doctors treating CJD patients will know to ask whether they ate venison.
CDC spokesman Ermias Belay says that the CDC will not be investigating the [Samii] cases because there is no evidence that the men ate CWD-infected meat. He notes that although the likelihood of CWD jumping the species barrier to infect humans cannot be ruled out 100% and that [we] cannot be 100% sure that CWD does not exist in humans& the data seeking evidence of CWD transmission to humans have been very limited.
Singeltary 2007
The Pathological Protein: Mad Cow, Chronic Wasting, and Other Deadly Prion Diseases
by Philip Yam
''Answering critics like Terry Singeltary, who feels that the US undercounts CJD, Schonberger _conceded_ that the current surveillance system has errors but stated that most of the errors will be confined to the older population''...
Revisiting Sporadic CJD
It’s not hard to get Terry Singeltary going. “I have my conspiracy theories,” admitted the 49-year-old Texan.1 Singeltary is probably the nation’s most relentless consumer advocate when it comes to issues in prion diseases. He has helped families learn about the sickness and coordinated efforts with support groups such as CJD Voice and the CJD Foundation. He has also connected with others who are critical of the American way of handling the threat of prion diseases. Such critics include Consumers Union’s Michael Hansen, journalist John Stauber, and Thomas Pringle, who used to run the voluminous www.madcow.org Web site. These three lend their expertise to newspaper and magazine stories about prion diseases, and they usually argue that
223
prions represent more of a threat than people realize, and that the government has responded poorly to the dangers because it is more concerned about protecting the beef industry than people’s health.
Singeltary has similar inclinations, but unlike these men, he doesn’t have the professional credentials behind him. He is an 11th-grade dropout, a machinist who retired because of a neck injury sustained at work. But you might not know that from the vast stores of information in his mind and on his hard drive. Over the years, he has provided unacknowledged help to reporters around the globe, passing on files to such big-time players as The New York Times, Newsweek, and USA Today. His networking with journalists, activists, and concerned citizens has helped medical authorities make contact with suspected CJD victims. He has kept scientists informed with his almost daily posting of news items and research abstracts on electronic newsgroups, including the bulletin board on www.vegsource.com and the BSE-listserv run out of the University of Karlsruhe, Germany. His combative, blunt, opinionated style sometimes borders on obsessive ranting that earns praise from some officials and researchers but infuriates others—especially when he repeats his conviction that “the government has lied to us, the feed industry has lied to us—all over a buck.” As evidence, Singeltary cites the USDA’s testing approach, which targets downer cows and examined 19,900 of them in 2002. To him, the USDA should test 1 million cattle, because the incidence of BSE may be as low as one in a million, as it was in some European countries. That the U.S. does not, he thinks, is a sign that the government is really not interested in finding mad cows because of fears of an economic disaster.
Singeltary got into the field of transmissible spongiform encephalopathy in 1997, just after his mother died of sporadic CJD. She had an especially aggressive version—the Heidenhain variant—that first causes the patient to go blind and then to deteriorate rapidly. She died just ten weeks after her symptoms began. Singeltary, who said he had watched his grandparents die of cancer, considered her death by CJD to be much, much worse: “It’s something you never forget.” Her uncontrollable muscle twitching became so bad “that it took three of us to hold her one time,” Singeltary recalled. “She did everything but levitate in bed and spin her head.” Doctors originally diagnosed Alzheimer’s disease, but a postmortem neuropathological exam demanded by Singeltary revealed the true nature of her death.
224 CHAPTER 14
Classifying a disease as “sporadic” is another way for doctors to say they don’t know the cause. Normal prion proteins just turn rogue in the brain for no apparent reason. The term “sporadic” is often particularly hard for the victims’ families to accept, especially when the patient was previously in robust health. Maybe it was something in the water, they wonder, or in the air, or something they ate—the same questions CJD researchers tried to answer decades ago. The names “sporadic CJD” and “variant CJD” also confuse the public and raise suspicions that U.S. authorities are hiding something when they say there have been no native variant CJD cases in the country.
Singeltary suspected an environmental cause in his mother’s demise—a feeling reinforced a year later when a neighbor died of sporadic CJD. For years, the neighbor had been taking nutritional supplements that contained cow brain extracts. Researchers from the National Institutes of Health collected samples of the supplement, Singeltary recounted, and inoculated suspensions into mice. The mice remained healthy—which only means that those supplement samples tested were prion-free.
Scientists have made several attempts during the past few decades to find a connection between sporadic CJD and the environment. Often, these studies take the form of asking family members about CJD victims—their diet, occupation, medical history, hobbies, pets, and so forth—and comparing them with non-CJD subjects. Such case-control CJD studies have produced some intriguing—and sometimes contradictory—results. In 1985, Carleton Gajdusek and his NIH colleagues reported a correlation between CJD and eating a lot of roast pork, ham, hot dogs, and lamb, as well as rare meats and raw oysters.2 Yet they also recognized that the findings were preliminary and that more studies were needed.
Following up, Robert Will of the U.K. National CJD Surveillance Unit and others pooled this data with those from two other case-control studies on CJD (one from Japan and one from the U.K.). In particular, they figured the so-called odds ratio—calculated by dividing the frequency of a possible factor in the patient group by the frequency of the factor in the control group. An odds ratio greater than 1 means that the factor may be significant. In their study, Will and his collaborators found an increase of CJD in people who have worked as health professionals (odds ratio of 1.5) and people who have had contact with cows
Laying Odds 225
(1.7) and sheep (1.6). Unfortunately, those connections were not statistically significant: The numbers of pooled patients (117) and control subjects (333) were so small that the researchers felt the odds ratios needed to reach 2.5 to 8 (depending on the assumptions) before they could be deemed statistically significant. The only statistically significant correlations they found were between CJD and a family history of either CJD (19.1) or other psychotic disease (9.9), although the latter might simply be correlated because psychotic disease may be an early symptom of undiagnosed CJD.3 In contrast with earlier findings, the team concluded that there was no association between sporadic CJD and the consumption of organ meats, including brains (0.6).
Although these case-control studies shed a certain amount of light on potential risk factors for CJD, it’s impossible to draw firm conclusions. Obtaining data that produces statistically meaningful results can be difficult because of the rarity of CJD and hence the shortage of subjects. Human memory is quite fragile, too, so patients’ families may not accurately recall the lifestyle and dietary habits of their loved ones over the course of a decade or more. Consequently, researchers must cope with data that probably contain significant biases. In a review paper on CJD, Joe Gibbs of the NIH and Richard T. Johnson of Johns Hopkins University concluded that “the absence of geographic differences in incidence is more convincing evidence against major dietary factors, since large populations eschew pork and some consume no meat or meat products.” A CJD study of lifelong vegetarians, they proposed, could produce some interesting data.4
The inconclusive results of case-control studies do not completely rule out the environment as a possible cause of CJD. “Dr. Prusiner’s theory does fit much of the data of spontaneous generation of [malformed] PrP somewhere in the brain,” Will remarked—that is, the idea that sporadic CJD just happens by itself falls within the realm of the prion theory. Still, “it’s very odd, if you look at all the forms of human prion diseases there are, all of them are transmissible in the laboratory and could be due to some sort of infectious agent.”5 One of the great difficulties, he explained, is that “given that this is a disease of an extraordinarily long incubation period, are we really confident that we can exclude childhood exposure that is transmitted from person to person, as people move around? It’s difficult to be sure about that.” There might a “carrier state” that leaves people healthy yet still able to
226 CHAPTER 14
infect others. If so, “you would never be able to identify what’s causing the spread of the disease,” concluded Will, who hasn’t stopped looking for a possible environmental link. He has some preliminary data based on studies that trace CJD victims’ lives well before the time symptoms began—up to 70 years; they suggest some degree of geographic clustering, but no obvious candidates for a source of infection.
A Case for Undercounting
The difficulty in establishing causal links in sporadic prion diseases—if there are any in the first place—underlines the importance of thorough surveillance. The U.K. has an active program, and when a victim of CJD is reported, one of Robert Will’s colleagues visits and questions the victim’s family. “No one has looked for CJD systematically in the U.S.,” the NIH’s Paul Brown noted. “Ever.”6 The U.S., through the Centers for Disease Control and Prevention, has generally maintained a more passive system, collecting information from death certificates from the National Center for Health Statistics. Because CJD is invariably fatal, mortality data is considered to be an effective means of tabulating cases. The CDC assessed the accuracy of such data by comparing the numbers with figures garnered through an active search in 1996: Teams covering five regions of the U.S. contacted the specialists involved and reviewed medical records for CJD cases between 1991 and 1995. Comparing the actively garnered data with the death certificate information showed that “we miss about 14 percent,” said CDC epidemiologist Lawrence Schonberger. “That’s improving. Doctors are becoming more knowledgeable,” thanks to increased scientific and media attention given to prion diseases.7
The active surveillance study of 1996, however, only looked at cases in which physicians attributed the deaths to CJD. Misdiagnosed patients or patients who never saw a neurologist were not tabulated— thus CJD may be grossly underreported. Many neurological ailments share symptoms, especially early on. According to various studies, autopsies have found that CJD is misdiagnosed as other ills, such as dementia or Alzheimer’s disease, 5 to 13 percent of the time. The CDC finds that around 50,000Americans die from Alzheimer’s each year
Laying Odds 227
(about 4 million have the disease, according to the Alzheimer’s Association). Therefore, one could argue that thousands of CJD cases are being missed. (On the flip side, CJD could be mistakenly diagnosed as Alzheimer’s disease or dementia, but the number of CJD patients is so small that they wouldn’t dramatically skew the statistics for other neurological ills.)
In part to address the issue of misdiagnosis, CJD families have asked the CDC to place the disease on the national list of officially notifiable illnesses, which tends to include more contagious conditions such as AIDS, tuberculosis, hepatitis, and viral forms of encephalitis. Currently, only some states impose this requirement. CDC officials have discounted the utility of such an approach, arguing that it would duplicate the mortality data, which is more accurate than early diagnoses of CJD, anyway. Moreover, mandatory reporting of CJD cases does not necessarily guarantee the end to missed cases.8
One clue suggests that the passive system is undercounting CJD in the U.S.: racial difference. The number of black CJD victims is about 38 percent that of white victims. Rather than sporadic CJD being a one in-a-million lottery, it’s more like one-in-2.5-million for African Americans. Access to medical care might be one reason. Schonberger recounted that the CDC had asked other countries with substantial black populations to submit CJD figures for comparison but found that the surveillance in those countries was inadequate. “We haven’t been able to find any comparable literature on this issue, so it’s still up in the air,” Schonberger said. On the other hand, Alzheimer’s disease is more common among black people than whites, with an estimated higher prevalence ranging from 14 percent to almost 100 percent, according to a February 2002 report by the Alzheimer’s Association. Are some black CJD cases being misdiagnosed as Alzheimer’s?
Answering critics like Terry Singeltary, who feels that the U.S. undercounts CJD, Schonberger conceded that the current surveillance system has errors but stated that most of the errors will be confined to the older population. As Schonberger pointed out, no doctor would misdiagnose a 30-year-old CJD patient as having Alzheimer’s. The average age of the first 100 variant CJD victims was 29; should the epidemiology of vCJD change—if older people start coming down with it—then there would be problems. “The adequacy of our overall CJD surveillance would be greatly reduced should the proportion of older individuals affected by variant CJD substantially increase,” Schonberger explained.9
SNIP...SEE FULL TEXT;
Singeltary Submission SEAC 2007
SEAC SPONGIFORM ENCEPHALOPATHY ADVISORY COMMITTEE Minutes of the 99th meeting held on 14th December 2007 Singeltary Submission
This was 22 years to the day Mom died from the Heidenhain Variant of Creutzfeldt Jakob Disease i.e. hvCJD, when i made this submission to SEAC and this was their reply to my questions of concern about cjd in the USA, my how things have changed...terry
SEAC SPONGIFORM ENCEPHALOPATHY ADVISORY COMMITTEE Minutes of the 99th meeting held on 14th December 2007
ITEM 8 – PUBLIC QUESTION AND ANSWER SESSION 40. The Chair explained that the purpose of the question and answer session was to give members of the public an opportunity to ask questions related to the work of SEAC. Mr Terry Singeltary (Texas, USA) had submitted a question prior to the meeting, asking: “With the Nor-98 now documented in five different states so far in the USA in 2007, and with the two atypical BSE H-base cases in Texas and Alabama, with both scrapie and chronic wasting disease (CWD) running rampant in the USA, is there any concern from SEAC with the rise of sporadic CJD in the USA from ''unknown phenotype'', and what concerns if any, in relations to blood donations, surgery, optical, and dental treatment, do you have with these unknown atypical phenotypes in both humans and animals in the USA? Does it concern SEAC, or is it of no concern to SEAC? Should it concern USA animal and human health officials?”
41. A member considered that this question appeared to be primarily related to possible links between animal and human TSEs in the USA. There is no evidence that sCJD is increasing in the USA and no evidence of any direct link between TSEs and CJD in the USA. Current evidence does not suggest that CWD is a significant risk to human health. There are unpublished data from a case of human TSE in the USA that are suggestive of an apparently novel form of prion disease with distinct molecular characteristics. However, it is unclear whether the case had been further characterised, if it could be linked to animal TSEs or if other similar cases had been found in the USA or elsewhere. In relation to the possible public health implications of atypical scrapie, H-type BSE and CWD, research was being conducted to investigate possible links and surveillance was in place to detect any changes in human TSEs. Although possible links between these diseases and human TSEs are of concern and require research, there is no evidence to suggest immediate public health action is warranted. The possible human health risks from classical scrapie had been discussed earlier in the meeting. Members noted that there are effective channels of discussion and collaboration on research between USA and European groups. Members agreed it is important to keep a watching brief on new developments on TSEs.
Heidenhain Variant Creutzfeldt Jakob Disease autopsy case report 'MOM'
DIVISION OF NEUROPATHOLOGY University of Texas Medical Branch 114 McCullough Bldg. Galveston, Texas 77555-0785
FAX COVER SHEET
DATE: 4-23-98
TO: Mr. Terry Singeltary @ -------
FROM: Gerald Campbell
FAX: (409) 772-5315 PHONE: (409) 772-2881
Number of Pages (including cover sheet):
Message:
*CONFIDENTIALITY NOTICE*
Snip…
FINAL AUTOPSY DIAGNOSIS
I. Brain: Creutzfeldt-Jakob disease, Heidenhain variant.
***TYPE: Anatomic(A) or Clinical(C)Diagnosis. IMPORTANCE: 1-immediate cause of death (COD); 2.ureterlying COD;3-contributory COD: 4.concomitant, significant; 5-incidental ***
Patient Name: POULTER, BARBARA Patient Location: AUTOPSY Room/Bed: Printed Date; Time: 01/30/98 - 0832
Page: 1 Continued ....
--------------
UTMB University of Texas Medical Branch Galveston, Texas 77555-0543 (409) 772-1238 Fax (409) 772-5683
Pathology Report
Autopsy NO,: AU-97-00435
MICROSCOPIC DESCRIPTION: The spongiform change is evident in all areas of neocortex, varying from mild to moderate in severity with only very mild neuronal loss and gliosis. In the bilateral occipital lobes, there is severe loss cortical neurons and gliosis, with a corresponding pallor of the underlying white matter. There is only minimal, focal spongiform change in corpus striatum, lentiform nuclei, thalamus, hippocampus, brainstem and cerebellum. There is no significant loss of neurons from the lateral geniculate nucleus, and the optic chiasm and tracts are well-myelinated.
SECTIONS TAKEN: N-l) Pituitary, N-2)Right frontal, N-3) Right inferior frontal, N-4) Right caudate putamen. N-5)Right lentiform nuclei, N-6) Right hippocampus, N-7) optic chiasm. N-8) Left inferior temporal lobe, N-9) Right inferior occipital, N-10} Cerabellum. N-l1)Midbrain, N-12) Pons, N-13) Medulla.
FINAL DIAGNOSES: BRAIN: 1. Clinical history of rapidly progressive dementia, clinically consistent with Creutzfeldt-Jakob Disease.
a. spongiform encephalopathy, most Severe in occipital lobes, consistent with Heidenhain variant of Creutzfeldt-Jakob disease.
b. Ventriculer enlargement, moderate, consistent with atrophy.
1. Communicating spherical enlargement of occipital horn of left lateral ventricle (possible incidental congenital anomaly).
DURA; Left subdural hemorrhage, recent, minimal.
PITUITARY: Severe capillary congestion.
COMMENTS; See also western blot report from Dr. Gambetti's lab Amyloid stains are not completed for this case as of this date. The results, which are not essential for the diagnosis, will be reported separately in an addendum.
(this was hand written notes) no amyloid evident in the special stains. no evidence of plaques. GAE
Gerald A. Campbell, M.D., Pathologist Division of Neuropathology
(Electronic Signature}. (Gross: 01/16/98Final: 02/08/98
Snip…
Page: 6 END OF REPORT
--------------
The University of Texas Medical Branch at Galveston
Gerald A, Campbell, Ph.D., M.D, Associate Professor and Director Division of Neuropathology Department of Pathology
February 26, 1998
Pierluigi Gambetti, M.D. Professor Institute of Pathology Case Western Reserve University 2085 Adelbert Road Cleveland Ohio 44106
Dear Dr, Gambetti:
Enclosed please find the microscopic slides and autopsy report from our patient, Barbara Poulter (Hosp.# 11851lQ,Autopsy # AU97-435). These slides are being sent for consultation at the request of Mr. Singletary, Ms. Poulter's son and next of kin. We will also send frozen tissue from the brain on dry ice next week, and someone will call you on the day the tissue is shipped. Please return the slides when you have finished with your examination. If you need any further information, please do not hesitate to call me. Thanks for your assistance with this case.
Sincerely, Gerald A. Campbell
------------------
CASE WESTERN RESERVE UNIVERSITY
February 26, 1988
Gerald Campbell, M.D,, PhD. Division of Neuropathology, G85 University TX Medical Branch Galveston, TX 77555-0785
Dear Dr. Campbell,
As per our telephone conversation concerning a recent case of CJD, I Will be willing to examine slides and the frozen tissue on western blotting, I will issue a report to you about our conclusions. Below is my address, Our Fed Ex number is XXXXXXXXXXXXXXX.
Thank your for your assistance in this matter,
Best personal regards,
Pierluigi Gambetti, M.D.
PG:In
Division of Neuropathology Pierluigi Gambetti, M.D. Director Institute Of Neuropathology 2085 Adelbert RoadCleveland, Ohio 44106
Phone 216-368-0587 Fax 216-368-2546
------------------
CASE WESTERN RESERVE UNIVERSITY
February 27, 1998
Dr. Gerald A. Campbell The University of Texas Medical Branch at Galveston Division of Neuropathology, G85 Galveston. TX77555-0785
Dear Dr. Campbell,
We are in receipt of the slides you senton Mrs. Barbara Poulter (your #: AU97-435;our#098-28).
Best personal regards, Pierluigi Gambetti, M.D.
PG:sb
Division of Neuropathology Pierluigi Gambetti, M.D., Director
-----------------------------------
CASE WESTERN RESERVE UNIVERSITY
March 30, 1998
Dr. Gerald A, Campbell The University of Texas Medical Branch at Galveston Division of Neuropathology Department of Pathology Galveston, Texas
Dear Dr Campbell,
We performed Western immunoblot analysis on the frozen tissue from your case #AU97-435 (our #098-28). The Immunoblot reveals the presence of protease-resistant prion protein (PrPres) confirming the diagnosis of prion disease. The immunoblot pattern of PrPres is consistent with the diagnosis of Creutzfeldt-Jakob disease.
Thank you for referring to us this interesting case.
Sincerely,
Piero Parchi, M.D.
Pierluigi Gambetti, M.D.
PP:sb
Division of Neuropathology Pierluigi Gambetti, M.D., Director Case Western Reserve University
This Autopsy report is for the use of anyone, who is trying to understand this hideous disease CJD. I hope it can be beneficial for some in researching human TSE. Please remember, this was my Mom, and to use this with great respect.
thank you, kind regards,
Terry S. Singeltary Sr., Bacliff, Texas USA
-------------------------------
BARBARA FREDERICK POULTER
DIED 12-14-97
If I had one last thing I could tell you, it would be, I love you. I'm sorry for the stupid argument we had the last few months, BEFORE this hideous disease ROARED through your body. BUT, I PROMISE MOM, YOUR DEATH WILL NOT GO UNANSWERED!
HEIDENHAN VARIANT CREUTZFELDT JAKOBDISEASE
We got a call from my Mother around the end of Oct. saying "the damn'est thing has happened, I can't see, and if I'm talking to you and I don't make sense, bare with me, I'll come back". It was a shock to all of us. It seems that a few days before, she was crossing the ferry and became frightened because she was having problems seeing. She explained it as looking down a tunnel or not being able to see from the sides, and seeing brown spots.
We had NOT been talking, over something, we had NO control of, for a few months. So I did not know she had been having these visual problems, until she was blind. These were her first symptoms. From that point on, I was with her most everyday.
I had to cross the Galveston/Bolivar ferry, and its about 30 minutes each way, so as the disease progressed, it gave me a great deal of time to think. When the visual problems started, it was about 2 weeks later, and she was blind. That led to coordination, and balance problems starting. But as this hideous disease progresses, it just GOES. You don't seem to catch up with it. It was like a fire in a hurricane. We would go out and get her things she needed one day, and the next day it would be obsolete, because the disease had gone to another stage. So you started over.
Her coordination and balancing led to being in a wheel-chair. She was starting to get these trembles. I also noticed how her hands and feet started to go inward. Her speech was nothing more than jerble at this time, and this was probably about the 6th week, (at this point we had to tie her to the wheel chair, to keep her from falling out). The trembles had turned into SEVERE JERKS, that at times would take 3 of us to hold her down. I will never forget that....About her 8th week she became comatose....She died around the 10th week. I had spent the night, she had problems through the night, so the nurse came. She checked her out and comforted us, (HOSPICE IS AWONDERFUL ORGANIZATION). The nurse said she seemed to be alright and that it would probably be alright to go home for a few hours. I was on the Ferry, going back to Galveston, when I got the call, she was gone. What can you do, Mom was gone, and I was stuck on the Damn Ferry, going the wrong direction.
She knew what she had. I remember, before she had lost her speech completely. After a doctors conference, and CJD had come up. She heard us say CJD, and she screamed, SHE knew! At that point, I didn't know what was, much less, CREUTZFELDT JAKOB DISEASE.....I have learned a lot since. I have learned I truly miss my Mom and I am MAD as hell that she is gone!
Terry/MADSON!!!
SYMPTOMS:
VISION - BLIND IN ABOUT 10 TO 14 DAYS
COORDINATION AND MUSCLE CONTROL, SWALLOWING DIFFICULTY, CONFUSION AND DEMENTIA, SPEECH PROBLEMS, HALLUCINATIONS, TREMBLES TOO SEVERE JERKING, LOSS OF WEIGHT, HANDS AND FEET GREW INWARD, UPPERTRUNK STIFFNESS, SHOULDER, UPPER ARM
CJD WATCH
CJD WATCH DATA BASE
CJD VOICE
CJD BLOOD/RECALL
CJD FOUNDATION
wasted days and wasted nights...Freddy Fender
terry
Terry S. Singeltary Sr., Bacliff, Texas, 77518, Galveston Bay, flounder9@verizon.net